Welche Routen der klinischen Bewertung von Medizinprodukten gibt es?
Bei medXteam stehen klinische Daten im Mittelpunkt. In diesem Kontext führen wir als CRO nicht nur klinische Prüfungen mit Medizinprodukten gemäß MDR und ISO 14155 durch, sondern bieten auch sämtliche weiteren Möglichkeiten und Formen der Datenerhebung und Produktzulassung sowie Marktüberwachung an. Im Fokus steht sowohl bei der Produktzulassung als auch im Rahmen der klinischen Nachbeobachtung immer die klinische Bewertung. Doch wie kann eine klinische Bewertung durchgeführt werden? Welche Möglichkeiten gibt es, die klinische Evidenz zu erbringen? Und welche Rolle spielen dabei die verschiedenen Routen der klinischen Bewertung? In diesem Blogbeitrag gehen wir diesen Fragen nach und erklären dabei insbesondere, was die drei Routen der klinischen Bewertung bedeuten, wann sie angewendet werden können und wie sie sich auf verschiedene Produktgruppen auswirken.
Abkürzungen
MDR Medical Device Regulation; EU-Verordnung 2017/745
PMCF Post-Market Clinical Follow-up, klinische Nachbeobachtung
CEP Clinical Evaluation Plan
CDP Clinical Development Plan
Zugrundeliegende Regularien
EU-Verordnung 2017/745 (MDR)
1. Einleitung
Die klinische Bewertung bildet einen essenziellen Schritt für jeden Hersteller von Medizinprodukten. Es ist erforderlich, für jedes Medizinprodukt einen umfassenden klinischen Bewertungsbericht (engl. Clinical Evaluation Report, CER) zu erstellen, der eine gründliche Literaturrecherche beinhaltet. Bereits vor dem Inkrafttreten der Verordnung (EU) 2017/745 (MDR) war dies Standardverfahren. Gemäß Artikel 61 der MDR ist daher die Planung und Durchführung einer klinischen Bewertung für sämtliche Medizinprodukte - von Klasse I bis Klasse III - vorgeschrieben:
„Der Hersteller legt den Umfang des klinischen Nachweises fest und begründet ihn, um die Erfüllung der relevanten grundlegenden Sicherheits- und Leistungsanforderungen nachzuweisen. Der Umfang des klinischen Nachweises muss den Merkmalen des Produkts und seiner Zweckbestimmung angemessen sein. Zu diesem Zweck führen Hersteller eine klinische Bewertung gemäß diesem Artikel und Anhang XIV Teil A durch, planen sie und dokumentieren sie.“ (Siehe Artikel 61 der MDR)
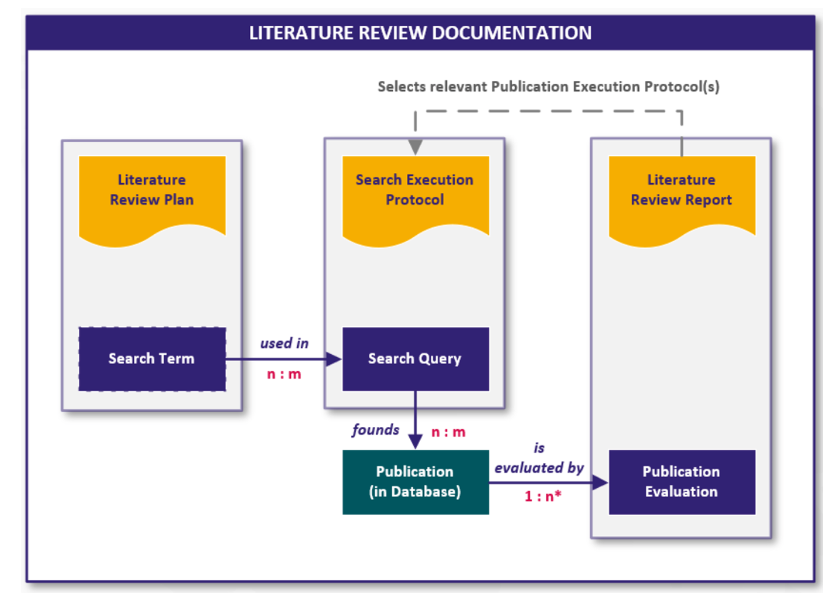
Dieser Prozess beginnt bereits früh im Entwicklungsprozess. Der Plan für die klinische Bewertung (engl. Clinical Evaluation Plan, CEP) wird in der Regel kurz nach der Festlegung der Produktidee, der Zweckbestimmung und der initialen Gefährdungsanalyse des Medizinprodukts erstellt.
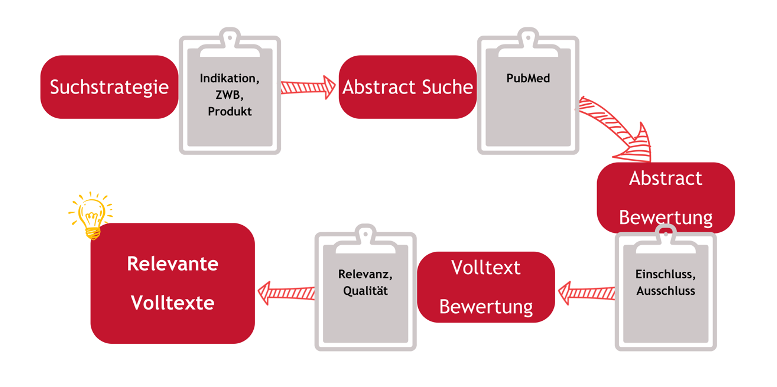
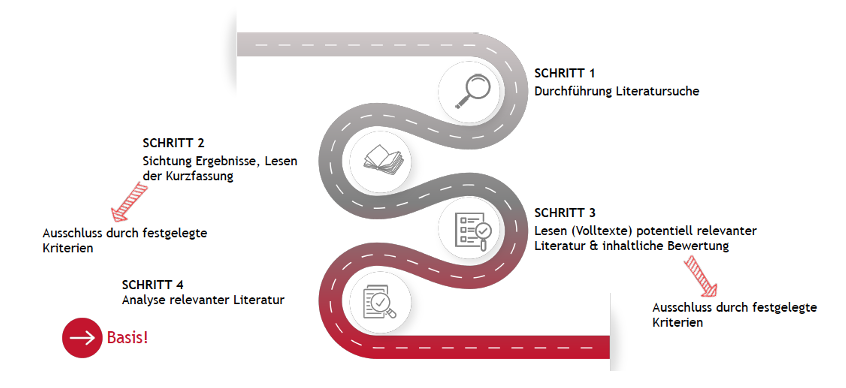
Während der Planung im CEP wird die Route festgelegt, welche Daten für die klinische Bewertung einbezogen werden sollen. Dazu gehören initiale Literaturrecherchen abhängig vom definierten Produkt sowie eine Marktbewertung in Bezug auf ähnliche Produkte und möglicherweise vorhandene klinische Daten in Publikationen und zum Stand der Technik im Anwendungsgebiet des Medizinprodukts.
Diese Informationen ermöglichen es, eine klinische Strategie für das Produkt festzulegen und diese im klinischen Entwicklungsplan (engl. Clinical Development Plan, CDP) festzuhalten.
Der frühe Zeitpunkt ist entscheidend, da die klinische Strategie und die daraus resultierende Route für die klinische Bewertung erheblichen Einfluss auf Zeit und Kosten des gesamten Entwicklungsprojekts haben. Es macht einen erheblichen Unterschied, ob eine klinische Prüfung noch in den Validierungspart integriert werden muss. Dies kann den Abschluss des Konformitätsbewertungsverfahrens und die CE-Kennzeichnung des Medizinprodukts um Jahre verzögern.
Die frühzeitige Planung ist auch deshalb wichtig, da sich dadurch die Zweckbestimmung noch ändern kann. Da diese die Grundlage des Entwicklungsprozesses bildet, können Änderungen zu einem fortgeschrittenen Zeitpunkt erhebliche Auswirkungen auf Zeit und Kosten des Projekts haben. (siehe hierzu auch unser Blog-Beitrag zur klinischen Strategie)
Daher sollte sich jeder Hersteller so früh wie möglich mit folgenden Fragen befassen:
Welche Produktklasse hat das Medizinprodukt? Für implantierbare Produkte der Klasse IIb und alle Klasse III Produkte ist nach unserer Erfahrung der Weg über eigene klinische Daten unumgänglich.
Wo liegt der Unterschied zu existierenden Produkten? Der Innovationsgrad des Produkts ist hier entscheidend.
Auf diese Fragen gibt nun dieser Blog-Beitrag die entsprechenden Antworten.
2. Die 3 Routen der klinischen Bewertung
Gemäß der MDR bezeichnet die klinische Bewertung einen strukturierten und geplanten Prozess zur fortlaufenden Generierung, Sammlung, Analyse und Bewertung von klinischen Daten eines Produkts, um dessen Sicherheit, Leistung und den klinischen Nutzen bei vorgesehener Verwendung durch den Hersteller zu überprüfen (MDR Art. 2, Satz 44). Klinische Daten werden wie folgt definiert:„Klinische Daten“ sind Informationen über die Sicherheit oder Leistung eines Produkts, die während seiner Anwendung gewonnen werden und aus verschiedenen Quellen stammen können (MDR Art. 2, Satz 48):
Klinische Studien des betreffenden Produkts.
- Klinische Studien oder andere Studien in der wissenschaftlichen Fachliteratur, die die Ähnlichkeit mit dem betreffenden Produkt nachweisen können.
- Berichte über klinische Erfahrungen mit dem Produkt oder ähnlichen Produkten, die nach dem Peer-Review-Verfahren in der wissenschaftlichen Fachliteratur veröffentlicht wurden.
- Klinisch relevante Informationen aus der Post-Marketing-Überwachung, einschließlich der klinischen Nachbeobachtung nach dem Inverkehrbringen.
Daraus ergeben sich drei mögliche Routen für die klinische Bewertung:
Eigene klinische Daten: Diese Route beinhaltet die Durchführung einer klinischen Studie mit dem betreffenden Produkt gemäß Artikel 62 der MDR, was eine sorgfältige Planung und Durchführung erfordert.
Klinische Daten zu äquivalenten Produkten: Hier werden klinische Daten zu ähnlichen Produkten aus der Fachliteratur verwendet oder es liegt bereits eine klinische Studie mit einem äquivalenten Produkt vor.
Verwendung von Leistungsdaten: Diese Route wird angewendet, wenn eine klinische Studie am Menschen nicht möglich oder sinnvoll ist. Stattdessen werden Leistungsdaten, auch Verifikationsdaten genannt, verwendet. Diese Daten basieren auf nichtklinischen Testmethoden, einschließlich Leistungsbewertung, technischer Prüfung und vorklinischer Bewertung.

Abb. 1 Die drei Routen der klinischen Bewertung
Es ist wichtig zu beachten, dass die dritte Route, obwohl in der MDR spezifiziert, bereits in der Richtlinie 93/42/EWG, MDD ähnlich festgelegt wurde. In den folgenden Abschnitten werden alle drei Routen im Detail beschrieben, wobei besonders auf die dritte Route eingegangen wird.
2.1 Eigene klinische Daten
Insbesondere für implantierbare Medizinprodukte der Klasse IIb stellt die Generierung eigener klinischer Daten unter der MDR die vorherrschende Methode dar. Während unter der Richtlinie 93/42/EWG die klinische Bewertung für diese Produkte noch über klinische Daten zu äquivalenten Produkten erfolgen konnte, ist dieser Ansatz unter den massiv verschärften Anforderungen der MDR nicht mehr möglich. Insbesondere die Voraussetzung, einen Vertrag mit dem Hersteller des potenziell äquivalenten Produkts abzuschließen, um vollständigen Zugang zu dessen technischer Dokumentation zu erhalten (MDR, Art. 61, Abschn. 5), schließt die Option der Nutzung äquivalenter Produkte vollständig aus:
„Ein Hersteller eines Produkts, das nachweislich einem bereits in Verkehr gebrachten, nicht von ihm hergestellten Produkt gleichartig ist, kann sich ebenfalls auf Absatz 4 berufen, um keine klinische Prüfung durchführen zu müssen, sofern zusätzlich zu den Anforderungen des genannten Absatzes die folgenden Bedingungen erfüllt sind: – Die beiden Hersteller haben einen Vertrag geschlossen, in dem dem Hersteller des zweiten Produkts ausdrücklich der uneingeschränkte Zugang zur technischen Dokumentation durchgängig gestattet wird, (…)“
Dieser Weg über eigene klinische Daten ist nicht nur für implantierbare Produkte der Klasse IIb und Klasse III obligatorisch, sondern auch für innovative Produkte mit klinischen Behauptungen zur Nutzen oder Wirksamkeit des Produkts. Für solche innovativen Produkte gibt es in der Regel keine äquivalenten Produkte, und die Route über Leistungs-/Verifizierungsdaten kann ebenfalls nicht gewählt werden, da klinische Behauptungen zwingend durch eigene klinische Daten nachgewiesen werden müssen.
Ein konkretes Beispiel wäre ein Produkt, dessen klinischer Nutzen die Reduktion von Schmerzen oder die Verbesserung der Lebensqualität ist. Die Wahl des Weges für die klinische Bewertung hängt hier vom Innovationsgrad des Produkts ab, unabhängig von dessen Klassifizierung. Dies kann sogar für Produkte der Klasse I gelten.
2.2 Die Äquivalenzroute
Unter der Richtlinie 93/42/EWG, MDD bzw. vor der Einführung der MDR galt die Äquivalenzroute als Standardverfahren – der sogenannte Goldstandard – für klinische Bewertungen. Wenn man jedoch klinische Daten zu einem äquivalenten Produkt nutzen möchte, um die Behauptungen zur Sicherheit, klinischen Leistung und klinischen Nutzen des eigenen Produkts zu untermauern, muss zunächst durch eine Literatursuche festgestellt werden, ob überhaupt klinische Daten zu diesem Produkt verfügbar sind. Ist dies nicht der Fall, ist eine obligatorische Äquivalenzbewertung nicht möglich. Liegen Daten zu diesem potenziellen Äquivalenzprodukt vor, dann wird in einem solchen Fall zunächst analysiert, ob das potenzielle Äquivalenzprodukt tatsächlich gleichwertig ist. Früher wurden für diese Analyse Bewertungskriterien verwendet, die bis zum Inkrafttreten der MDR im Mai 2021 im Leitfaden MEDDEV 2.7/1 Rev. 4.3 für klinische Bewertungen festgehalten waren.
Diese Kriterien zielten auf die klinischen, technischen und biologischen Eigenschaften des Äquivalenzprodukts ab, die mit dem eigenen Produkt verglichen wurden, um festzustellen, ob sie in einigen Aspekten gleich oder nur ähnlich sind. Beispielsweise mussten sie möglicherweise für dieselben Indikationen eingesetzt werden (klinische Merkmale), während technische Merkmale wie Durchmesser und Größe ähnlich sein konnten.
Mit der Einführung der MDR und des zugehörigen MDCG-Dokuments 2020-05 („Clinical Evaluation – Equivalence: A guide for manufacturers and notified bodies“) wurden diese Kriterien drastisch verschärft. Insbesondere im Hinblick auf die technische und biologische Äquivalenz müssen die Produkte in ihren Merkmalen nun deutlich häufiger identisch sein als zuvor. Beispielsweise erfordert die Bewertung der Äquivalenz bei einer Software als Medizinprodukt möglicherweise den Zugang zu vollständigen Algorithmen und Source Codes der anderen Software, wobei diese Merkmale dann identisch sein müssten. Bei stofflichen Medizinprodukten müssen beide Produkte aus genau denselben Stoffen bestehen und in derselben Konzentration vorliegen, und auch die Produktrückstände müssen identisch sein.
Solche detaillierten Daten zum potenziellen Äquivalenzprodukt liegen in der Regel nicht vor, da niemand Zugriff auf solche Details einer Software oder exakten stofflichen Konzentrationen und Rückstände eines Produkts hat. Und genau das erschwert zunehmend die Äquivalenzroute bzw. macht sie gar unmöglich.
2.3 Leistungsdaten
Der Weg, die klinische Leistung eines Produkts über Leistungsdaten nachzuweisen, war schon immer möglich und bleibt es auch weiterhin unter der MDR (Artikel 61):
„Wird der Nachweis der Übereinstimmung mit grundlegenden Sicherheits- und Leistungsanforderungen auf der Grundlage klinischer Daten für ungeeignet erachtet, ist jede solche Ausnahme auf der Grundlage des Risikomanagements des Herstellers und unter Berücksichtigung der besonderen Merkmale des Zusammenspiels zwischen dem Produkt und dem menschlichen Körper, der bezweckten klinischen Leistung und der Angaben des Herstellers angemessen zu begründen; dies gilt unbeschadet des Absatzes 4. In diesem Fall muss der Hersteller in der technischen Dokumentation gemäß Anhang II gebührend begründen, warum er den Nachweis der Übereinstimmung mit grundlegenden Sicherheits- und Leistungsanforderungen allein auf der Grundlage der Ergebnisse nichtklinischer Testmethoden, einschließlich Leistungsbewertung, technischer Prüfung („bench testing“) und vorklinischer Bewertung, für geeignet hält.“
Die Entscheidung basiert auf verschiedenen Überlegungen:
- dem Ergebnis des Risikomanagements
- den Besonderheiten der Wechselwirkung zwischen Körper und Produkt
- dem Nachweis der Leistungsfähigkeit basierend auf Produktprüfungen (technisch, in-vitro)
- dem Ergebnis der vorklinischen Bewertung (initiale Literaturrecherche, Verifizierungstests usw.)
Diese Entscheidung muss im Plan für die klinische Bewertung angemessen begründet und dokumentiert werden.
Diese Route wird gewählt, wenn eine klinische Prüfung wenig Sinn ergibt. Ein klassisches Beispiel hierfür ist der Mundspatel aus Holz, für den klinische Daten in der Literatur nicht verfügbar sind. In solchen Fällen belegen technische Daten wie Bruchfestigkeit und Verarbeitung die Sicherheit und Leistungsfähigkeit des Produkts.
Obwohl diese Route in der Vergangenheit weniger genutzt wurde, da sie oft weniger bekannt war und üblicherweise die Route über ein Äquivalenzprodukt genutzt wurde, ist sie für eine Vielzahl von Produkten geeignet.
2.3.1 Beispiel – Medizinische Software
Die meisten Software-Produkte (Klasse I und IIa) sind Beispiele für Produkte, bei denen der Weg über Leistungsdaten sinnvoll ist. Die Begründung für diese Entscheidung ist wie folgt:
Das Produkt wurde im Rahmen des Software-Lebenszyklusprozesses gemäß IEC 62304 umfassend verifiziert, und alle Tests wurden erfolgreich abgeschlossen. Die Tests umfassten Unit-Tests, Integrationstests, Systemtests und Usability-Tests. Basierend auf diesen Tests kann gezeigt werden, dass das Produkt effektiv funktioniert.
Gemäß MDCG-2020-1 (Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software) wird die wissenschaftliche Validität als das Ausmaß definiert, in dem der Output des Software-Produkts auf der Grundlage der ausgewählten Inputs und Algorithmen mit dem angestrebten physiologischen Zustand oder der klinischen Erkrankung assoziiert ist. Um den Nachweis der wissenschaftlichen Validität zu erbringen, wird eine Literatursuche durchgeführt, die auch den Nachweis des Nutzens gemäß der MDR sowie die Ermittlung des State-of-the-Arts und die Identifizierung der Sicherheit und Leistungsfähigkeit des Medizinprodukts beinhaltet.
Die klinisch relevanten Komponenten des Systems sind die Implementierungen der Algorithmen/Fragebögen zur Diagnose oder zum Therapieverlauf. Die Literaturrecherche konzentriert sich auf Scores/Erkennungsalgorithmen sowie auf den allgemeinen Einsatz digitaler Produkte in der Diagnose/Therapie der genannten Indikationen.

Tabelle 1: Klinische Bewertung eines Softwareproduktes
2.3.2 Beispiel – Zahnarztstuhl
Ein weiteres Produkt, dessen klinische Leistung, Sicherheit und Nutzen gut über Leistungsdaten bewertet werden können und für das eine klinische Prüfung keinen Sinn ergibt, ist die dentale Behandlungseinheit: der Zahnarztstuhl.
Solche Produkte sind aktive Medizinprodukte, die zur Behandlung von Kindern und Erwachsenen im zahnmedizinischen Bereich dienen. Diese Produkte sind zahnärztliche Behandlungsgeräte nach ISO 7494 mit einem zahnärztlichen Patientenstuhl nach ISO 6875. Sie sind ausschließlich für den Einsatz in der Zahnheilkunde vorgesehen und dürfen nur von medizinischem Fachpersonal bedient werden. Die dentale Behandlungseinheit wird als Hilfsmittel zur Patientenlagerung und zur Behandlung im dentalmedizinischen Bereich eingesetzt. Abhängig davon, ob Dentalinstrumente Teil dieser Behandlungseinheit sind und wenn ja, welche, werden diese Produkte in die Klasse IIa oder IIb eingestuft.
Aufgrund der klaren Zweckbestimmung dieser Produkte erübrigt sich die Frage, ob eine klinische Prüfung am Menschen durchgeführt werden soll. Die Behauptungen zum Produkt beziehen sich auf die Ergonomie sowohl für den Patienten als auch für den Behandler und Anwender des Produkts. Außerdem wird ein effizientes und einfaches Arbeiten hervorgehoben, und vorgeschriebene Verfahren sowie unterstützende Komponenten dienen der Erleichterung der Infektionskontrolle und der Aufrechterhaltung der Wasserqualität. Diese Aussagen sind keine geeigneten Endpunkte für eine klinische Prüfung. Sie können jedoch mit Leistungsdaten belegt werden. Zum Beispiel kann das Thema Ergonomie und einfache Anwendung über den Test zur Gebrauchstauglichkeit (DIN EN 62366-1) belegt werden. Die Einhaltung der jeweiligen Normen und Vorschriften zur Wasserhygiene und -qualität bestätigt ebenfalls diese Behauptungen zum Produkt. Die Begründung für die Wahl des Weges über Leistungsdaten ist hier nun in Tabelle 2 aufgeführt:

Tabelle 2: Klinische Bewertung eines aktiven Produkts
2.3.3 Beispiel – Herzrhythmus Detektor
Ein weiteres Beispiel ist ein Klasse IIa-Produkt, das Episoden von unregelmäßigem Herzrhythmus, die auf Vorhofflimmern hinweisen, durch Langzeitüberwachung der Pulsparameter über mehrere Tage bis zu vier Wochen erkennen kann. Es unterstützt somit die Diagnose, indem es Hinweise auf Vorhofflimmern liefert.
Diesem Produkt liegt eine eingebettete Software zugrunde, über deren Algorithmus die Episoden erkannt und entsprechend angezeigt werden. Die Verifizierung und Validierung der Software liefert bereits entscheidende Daten zur Funktionsweise dieses Medizinprodukts. Trotz der Möglichkeit, eine klinische Prüfung am Menschen durchzuführen, müssen auch ethische Bedenken berücksichtigt werden. Eine Ethikkommission prüft genau diese Aspekte. Es gibt jedoch alternative Wege, um klinische Daten zur Erfüllung der klinischen Leistung und Funktion des Produkts zu generieren. Beispielsweise können Episoden über Simulationstests eingespielt werden, um zu überprüfen, ob der Algorithmus sie korrekt erkennt. Auch hier bedarf es keiner Humanstudie, um diesen Nachweis zu erbringen. Die Begründung für diese Route ist in der folgenden Tabelle aufgeführt:

Tabelle 3: Klinische Bewertung Herzrhythmus-Detektor
2.3.4 Beispiel – Dentalimplantat
Selbst mit einem implantierbaren Produkt kann dies ein gangbarer Weg sein, wie unser letztes Beispiel aus der Zahntechnik zeigt: Die Titanbasis ist ein Teil eines Dentalimplantats, ein implantierbares Medizinprodukt der Klasse IIb. Die Titanbasis dient der Herstellung eines individuell angefertigten implantatprothetischen Aufbaus. Sie stellt nach dem Verkleben mit einem CAD/CAM gefrästen Aufbau das Verbindungselement zum Implantat dar. Sie kann auch einzeln vertrieben werden, sodass auch für dieses Produkt eine klinische Bewertung erstellt werden muss.
Bei der Durchführung einer Literatursuche im Bereich der Dentalimplantate stößt man schnell auf die Grenzen solcher Systemkomponenten. Denn es gibt noch keine Humanstudie, die ausschließlich die Titanbasis als Prüfprodukt untersucht hat. Publiziert wurden lediglich In-vitro-Studien oder Studien zu Materialeigenschaften (Titan) usw. Wie die Wahl der Route über Leistungsdaten in diesem Fall begründet wird, ist in Tabelle 4 aufgeführt:

Tabelle 4: Klinische Bewertung Titanbasis
2.3.5 Fazit aus den Beispielen
Bei all diesen Beispielen hat auch der Abschnitt zur klinischen Bewertung des Standes der Technik einen hohen Stellenwert. Viele Produkteigenschaften oder -funktionen und in vielen Fällen auch der klinische Nutzen lassen sich über Leitlinien, technische Dokumente sowie Standards belegen. Was bei diesen Beispielen ebenfalls unterstützend hinzukommt, ist die Datenerhebung im Rahmen der klinischen Nachbeobachtung (Post-Market Clinical Follow-up, PMCF) – nachdem das Produkt in Verkehr gebracht wurde und das CE-Kennzeichen trägt. Aus einer klinischen Bewertung, die auf Leistungsdaten fußt, resultieren in der Regel Maßnahmen im Rahmen der klinischen Nachbeobachtung. Diese können von fokussierten Literatursuchen über Produktregister bis hin zu Anwendungsbeobachtungen und PMCF-Studien reichen. Damit lassen sich gezielt Lücken schließen, die über die Leistungsdaten noch nicht vollumfassend belegt werden konnten. Eine solche Vorgehensweise wird bei einer korrekten Begründung auch von den Benannten Stellen anerkannt und akzeptiert.
3. Schlussfolgerung
Bisher wurden für viele Medizinprodukte die Äquivalenzroute und die Nutzung klinischer Daten zu einem oder mehreren Äquivalenzprodukten unabhängig von der Klasse des Medizinprodukts gewählt. Mit dem Inkrafttreten der MDR hat sich dies jedoch vollständig geändert. Aufgrund der strengeren Regulierungen, insbesondere für implantierbare und Klasse III Produkte, ist diese Route kaum noch möglich. Dies liegt sowohl an der erschwerten Nachweisführung der Äquivalenz als auch an den konkreten Vorschriften, wie dem Vertragsabschluss zwischen den Herstellern (MDR, Art. 61 Abschn. 5). Diese Veränderung dürfte wahrscheinlich auch das angestrebte Ziel der Macher der MDR gewesen sein.
Daher ist es entscheidend, bereits zu Beginn des Entwicklungsprozesses initiale Literaturrecherchen durchzuführen und die klinische Strategie zu überdenken. Dies ermöglicht eine umfassende Betrachtung der Datensituation und des Standes der Technik zum Produkt. Eine frühzeitige Festlegung der Zweckbestimmung kann dazu führen, dass der Weg über Leistungsdaten eingeschlagen werden kann, was nun an Bedeutung gewinnt und bei immer mehr Produkten Anwendung findet. Die Beispiele in diesem Beitrag zeigen, dass dies möglich ist, wenn es fundiert begründet werden kann. Dennoch darf auch bei einer klinischen Bewertung basierend auf Leistungsdaten eine Literatursuche nicht vernachlässigt werden. Daten zum Stand der Technik, Leitlinienempfehlungen sowie technische Standards tragen hier maßgeblich zur Beurteilung bei.
4. Wie wir Ihnen helfen können
Als CRO unterstützen wir Sie über den gesamten Prozess der Generierung und Bewertung klinischer Daten und bei der Zulassung und Marktüberwachung Ihres Produkts. Und dabei beginnen wir mit der klinischen Strategie! Außerdem erstellen wir die komplette klinische Bewertungsakte für Sie.
Im Falle von klinischen Prüfungen überlegen wir gemeinsam mit Ihnen, ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss. Das klären wir im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung.
Wenn eine klinische Prüfung durchgeführt werden soll, müssen zuvor grundlegende Sicherheits- und Leistungsanforderungen erfüllt sein. Die Daten aus der klinischen Prüfung münden dann in die klinische Bewertung, die wiederum die Basis für Post-Market-Clinical-Follow-up (PMCF)-Aktivitäten (ggf. einschließlich einer PMCF-Studie) darstellt.
Außerdem benötigen alle Hersteller von Medizinprodukten ein Qualitätsmanagement system (QMS), auch bei der Entwicklung von Produkten der Klasse I.
Wir unterstützen Sie während Ihres kompletten Vorhabens mit Ihrem Medizinprodukt, beginnend bei einer kostenlosen Erstberatung, Hilfe bei der Einführung eines QM Systems, Studienplanung und Durchführung bis hin zur Technischen Dokumentation - immer mit primärem Bezug auf die klinischen Daten zum Produkt: von Anfang an bis zum Ende.
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung