Im ersten Blog-Beitrag nach der Sommerpause schieben wir die neuen MDCG-Dokumente ein, die konkret klinische Prüfungen mit Medizinprodukten betreffen. In diesem Beitrag stellen wir beide Dokumente vor und gehen auf die Punkte ein, die für Sponsoren und somit auch die Hersteller von Belang sein können.
Abkürzungen
CE Marking on a product to signify that it meets the legal requirements to be sold on the extended Single Market in the European Economic Area (EEA).
CIP Clinical investigation plan
DMIDS Deutsches Medizinprodukte-Informations- und Datenbanksystem
EUDAMED European database on medical devices
GSLA Grundlegende Sicherheits- und Leistungsanforderungen
IVDR Verordnung (Eu) 2017/746 des Europäischen Parlaments und des Rates über In-Vitro-Diagnostika
MDCG Medical Device Coordination Group
MDR Medical Device Regulation; EU-Verordnung 2017/745
MPI Medizinprodukteinformationssystem
MS Member State
NCA National Competent Authority
PMCF Post-market clinical follow-up
REC Research ethics committee
Zugrundeliegende Regularien
EU-Verordnung 2017/745 (MDR)
MDCG 2021-6
MDCG 2021-8
1. Einleitung
Die Medical Device Coordination Group (MDCG) veröffentlicht regelmäßig und kontinuierlich sogenannte Guidance-Dokumente zur MDR. Dies erfolgt gemäß Artikel 105 der MDR und Artikel 99 der IVDR. Die Dokumente werden in Zusammenarbeit mit interessierten Parteien, die in den verschiedenen Gruppen vertreten sind, verfasst und sind in folgendem Format angegeben: "MDCG Jahr-Nummer-Revision".
Die MDCG setzt sich aus Vertretern aller Mitgliedstaaten zusammen, und ein Vertreter der Europäischen Kommission führt den Vorsitz. Die Dokumente sind keine Dokumente der Europäischen Kommission und können nicht als offizieller Standpunkt der Europäischen Kommission angesehen werden. Alle in diesem Dokument geäußerten Ansichten sind rechtlich nicht bindend, und nur der Gerichtshof der Europäischen Union kann eine verbindliche Auslegung des Unionsrechts vornehmen.
Sie stellen lediglich eine gemeinsame Auffassung darüber dar, wie die MDR und die IVDR in der Praxis angewendet werden sollten, damit eine wirksame und harmonisierte Umsetzung der Rechtsvorschriften erreicht werden kann.
Dieser Beitrag beschäftigt sich nun mit den beiden neu veröffentlichten MDCG-Dokumenten 2021-6 und 2021-8.
Dabei richtet sich das Dokument MDCG 2021-6 „Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation“ an Sponsoren von klinischen Prüfungen von Medizinprodukten, die im Geltungsbereich der Verordnung (EU) 2017/745 (MDR) durchgeführt werden. Es behandelt Fragen und Antworten und kann zu gegebener Zeit durch weitere Fragen und Antworten ergänzt werden.
Das Dokument MDCG 2021-8 „Clinical investigation application/notification documents” adressiert dagegen die beim Antrag auf eine klinische Prüfung einzureichenden Dokumente.
2. Neue MDCG-Dokumente
2.1 MDCG 2021-6
Insgesamt 28 allgemeine Fragen und zugehörige Antworten umfasst dieses MDCG-Dokument, das im vergangenen April veröffentlicht wurde:
Dabei wird zum einen auf die Unterschiede zwischen der MDR und der vorherigen Richtlinie 93/42/EWG eingegangen:
What are the general differences and improvements related to clinical investigations under the new Regulation (EU) 2017/745 (MDR) as compared to the Directives 93/42/EEC and 90/385/EEC?
Im Grunde gibt es hier keine, außer dass die MDR ein anderes, verbindlicheres Regelwerk darstellt, anhand dessen eine einheitlichere und vorhersehbarere Durchführbarkeit von klinischen Prüfungen gewährleistet werden soll.
Neben der Definition der klinischen Prüfung, die der MDR entnommen wurde (Frage 2) ist die dritte Fragestellung wieder interessant:
What is the difference between the performance, clinical performance and clinical benefit?
Gemäß der MDR ist die Leistung (Definition s. Artikel 2(22) der MDR) eines Produkts die Fähigkeit des Produkts, seine vom Hersteller angegebene Zweckbestimmung zu erfüllen. Im weiteren Sinne ist die klinische Leistung (Definition s. Artikel 2(52) der MDR) eines Medizinprodukts die Fähigkeit des Produkts, seine Zweckbestimmung zu erfüllen und dadurch bei bestimmungsgemäßem Gebrauch einen klinischen Nutzen (Definition s. Artikel 2(53) der MDR) zu erbringen. Der klinische Nutzen ist die positive Auswirkung eines Produkts auf die Gesundheit einer Person, ausgedrückt in Form eines aussagekräftigen, messbaren, patientenrelevanten klinischen Ergebnisses (einschließlich diagnosebezogener Ergebnisse) oder einer positiven Auswirkung auf das Patientenmanagement oder die öffentliche Gesundheit.
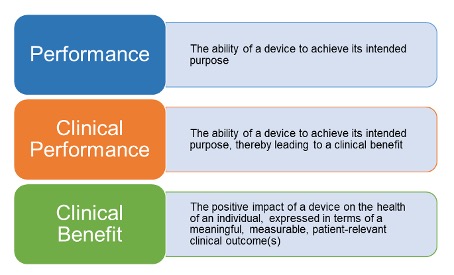
Abb. 1 Definition von Leistung, klinischer Leistung und dem klinischen Nutzen.
Bisher wurde nie so exakt nach Leistung (zur Erfüllung der Zweckbestimmung) und klinischer Leistung (Leistung zur Erbringung des klinischen Nutzens, also die klinischen Aspekte der Leistung) unterschieden.
Das ist aber wichtig, nicht nur in Bezug auf klinische Prüfungen bei der Wahl der primären und sekundären Endpunkte, sondern auch beim Festlegen der Claims (Behauptungen) eben zur klinischen Leistung im Rahmen der klinischen Bewertung.
Die neunte Frage umfasst das Thema „zusätzliche belastende Untersuchungen“ und definiert diese im Einklang mit Artikel 74.
„Zusätzliche Verfahren, die eine Belastung darstellen, können eine Vielzahl unterschiedlicher Eingriffe umfassen, darunter Verfahren, die Schmerzen, Unbehagen, Angst, potenzielle Risiken oder Komplikationen/Nebenwirkungen, Störungen des Lebens und persönlicher Aktivitäten oder andere unangenehme Erfahrungen verursachen können. Sie wird meist aus der Perspektive der Person bestimmt, die die Belastung trägt.
Weitere invasive Verfahren sind unter anderem das Eindringen in das Körperinnere durch die Körperoberfläche, auch durch die Schleimhäute von Körperöffnungen, oder das Eindringen in eine Körperhöhle durch eine Körperöffnung.“
Dazu zählen beispielsweise bereits Blutabnahmen oder auch Biopsien, die zusätzlich im Rahmen der klinischen Prüfung durchgeführt werden und nicht zum klinischen Routinealltag gehören.
Hier herrscht jedoch noch Unsicherheit und es empfiehlt sich im Rahmen der jeweiligen klinischen Prüfung, die Ethikkommission und auch die Behörde zu kontaktieren, um bei ggf. strittigen Fragestellungen bzgl. der zusätzlichen Belastung die richtige Entscheidung hinsichtlich Antrag treffen zu können.
Insgesamt beschäftigen sich viele Fragen mit dem korrekten regulatorischen Weg, der in der jeweiligen Konstellation des Medizinproduktes bzgl. einer klinischen Prüfung beschritten werden muss. Hier geht es um die Artikel 62, 74 und auch 82 der MDR. Hierzu beraten wir individuell gerne im Rahmen unserer kostenlosen Erstberatung.
Ab Frage 15 bis einschließlich Frage 20 wird auf Änderungen des klinischen Prüfplans (Amendments) eingegangen und die Fragen 21 bis 28 befassen sich mit Fristen und Übergangsfristen.
Als grundsätzliche Übersicht ist dieses MDCG-Dokument sicher geeignet, um grundsätzliche Fragestellungen kurz anzureißen. Detaillierte Hinweise bzw. Erklärungen sind aber nicht zu finden und müssen im Rahmen der Beratung in Bezug auf den Einzelfall geklärt werden.
2.2 MDCG 2021-8
Eine klinische Prüfung muss außer im Falle einer PMCF klinischen Prüfung, die nicht unter Artikel 74 der MDR fällt, mit einem Antrag mit den in Anhang XV Kapitel II der MDR genannten Unterlagen über das in Artikel 73 der MDR genannte elektronische System eingereicht werden.
Da es die europäische Datenbank für Medizinprodukte (EUDAMED) noch nicht gibt, hat nun die MDCG eine Reihe von Dokumenten für die Beantragung/Meldung klinischer Prüfungen erstellt, um die Verfahren für klinische Prüfungen im Rahmen der MDR zu unterstützen.
Achtung: In Deutschland werden die klinischen Prüfungen über das Medizinprodukteinformationssystem, nun Deutsches Medizinprodukte-Informations- und Datenbanksystem (DMIDS) bei BfArM (früher DIMDI) beantragt, das die im MDCG-Dokument aufgeführten Templates als Abfragen bereits enthält.
Zu diesen Dokumenten gehören:
- Klinische Prüfung - Antrag/Meldeformular im Rahmen der MDR
- Addendum zum Antrag/Meldeformular für klinische Prüfungen für:
- Zusätzliche(s) Prüfprodukt(e) (Abschnitt 3)
- Zusätzliche(s) Vergleichspräparat(e) (Abschnitt 4)
- Zusätzliche Prüfstelle(n) (Abschnitt 5)
- Unterstützende Dokumente für die klinische Prüfung - Anhang der beizufügenden Dokumente
- Checkliste der allgemeinen Sicherheits- und Leistungsanforderungen, Normen, gemeinsamen Spezifikationen und wissenschaftlichen Empfehlungen
Soweit möglich, enthält das Antrags-/Meldeformular für die klinische Prüfung die gleichen Datenfelder wie das sich noch in der Entwicklung befindliche EUDAMED-System.
Die Templates sind im MDCG-Dokument aufgeführt und über folgende Links zu erreichen:
Clinical investigation – application form under Medical Device Regulation
Additional investigational device(s) (section 3)
Additional comparator device(s) (section 4)
Additional investigation site(s) (section 5)
Außerdem enthält dieses Dokument dann noch einen Link zu einer Auflistung der benötigten Dokumente, die sich an der ISO 14155:2020 ausrichtet.
Schließlich erhält man ein Template zur Checkliste der grundlegenden Sicherheits- und Leistungsanforderungen (GSLA), Normen, Common Specifications usw., das sich an der MDR-Checkliste anlehnt:
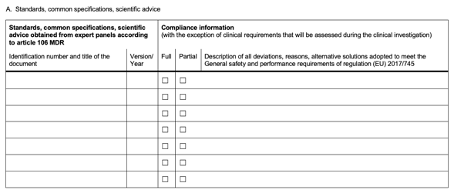
Abb. 2 Auflistung der Normen, CS und wissenschaftlichen Hinweise, außer jenen, die im Rahmen der klinischen Prüfung untersucht werden
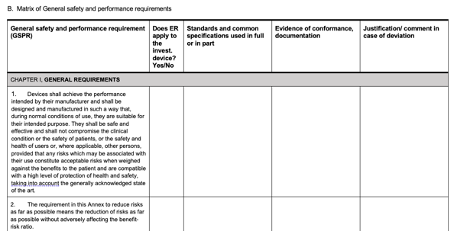
Abb. 3 Matrix zur Erfüllung der GSLA
Bis auf die beiden Word-Templates liefert dieses MDCG-Dokument für klinische Prüfungen, die in Deutschland durchgeführt werden, keine neuen Informationen, da hier ja über das DMIDS alles entsprechend eingetragen und beantragt werden kann.
3. Was wir für Sie tun können
Wir fungieren als wissenschaftliche herstellerunabhängige Institution (CRO). Als solche unterstützen wir Sie während Ihrem kompletten Vorhaben bzgl. klinischer Prüfung – von der ersten Idee bis zur Auswertung und dem klinischen Prüfbericht.
4. Wie wir Ihnen helfen können
Ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss, klären wir bei medXteam im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung.
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung