Die Blog-Reihe geht in diesem Jahr in der Adventszeit mit unserem diesjährigen dreiteiligen „Adventsspezial“ weiter. Hiermit möchten wir Sie umfassend über die wichtigen Schnittstellen bei den verschiedenen Typen der klinischen Prüfungen mit der technischen Dokumentation und den jeweiligen Dokumenten informieren.
Das Besondere an unserer Aktion ist dabei, dass der Beitrag sich über die ersten drei Adventswochen aufteilt. In jeder Woche werden ein Typ der klinischen Prüfungen und die jeweiligen Schnittstellen detailliert betrachtet. Im Januar wird es dann mit dem Thema Risikomanagement bei klinischen Prüfungen weitergehen.
Der erste Teil unseres Blog-Spezials befasste sich mit den Schnittstellen bei der „Zulassungsstudie“ (klinische Prüfungen gemäß Artikel 62 der MDR). Dieser zweite Teil 2 beleuchtet nun die PMCF-Studien (Artikel 74 der MDR, MPDG, ISO 14155). Der dritte und letzte Teil erklärt nächste Woche die Schnittstellen bei DiGA-Studien.
Abkürzungen.
BOB (Bundesoberbehörde)
EK (Ethikkommission)
KP (klinische Prüfung)
MDR (medical device regulation; Verordnung 2017/745)
MPEUAnpG (das Medizinprodukte-EU-Anpassungsgesetz wurde am 25.05.2020 vom Bundestag als Gesetze verabschiedet. Dieses MPAnpG-EU beschreibt im Artikel 1 das Medizinprodukte-Durchführungsgesetz (MPDG))
MPDG (das MPDG wird das Medizinproduktegesetz (MPG) ab 26. Mai 2021 schrittweise ablösen und für alle Hersteller und Betreiber von Medizinprodukten in Deutschland rechtsverbindlich sein).
Teil 2: Schnittstellen zur technischen Dokumentation bei PMCF-Studien und Verbindung zur MDR
1. Einleitung
Der korrekte Begriff für eine klinische Studie mit Medizinprodukten ist „klinische Prüfung“.
Man unterscheidet die folgenden Typen von klinischen Prüfungen:
- Grundlagenforschung: sonstige klinische Prüfung (MDR Artikel 82)
- Pilotstudie/Zulassungsstudie: klinische Prüfungen zum Nachweis der Konformität von Produkten (MDR Artikel 62)
- PMCF-Studie: klinische Prüfungen in Bezug auf Produkte, die die CE-Kennzeichnung tragen (MDR Artikel 74)
Hinzu kommt nun noch speziell in Deutschland die sog. DiGA-Studie:
- Studie mit einer digitalen Gesundheitsnwendung (DiGA) zum Nachweis positiver Versorgungseffekte zur Erlangung eines Erstattungsstatus.
- d. R. mit CE-gekennzeichnetem Medizinprodukt: PMCF-Studie
- bei Einplanung in Zulassungsprozess auch Zulassungsstudie möglich
(Quellen: DiGAV, DVG, DiGA-Leitlinie)
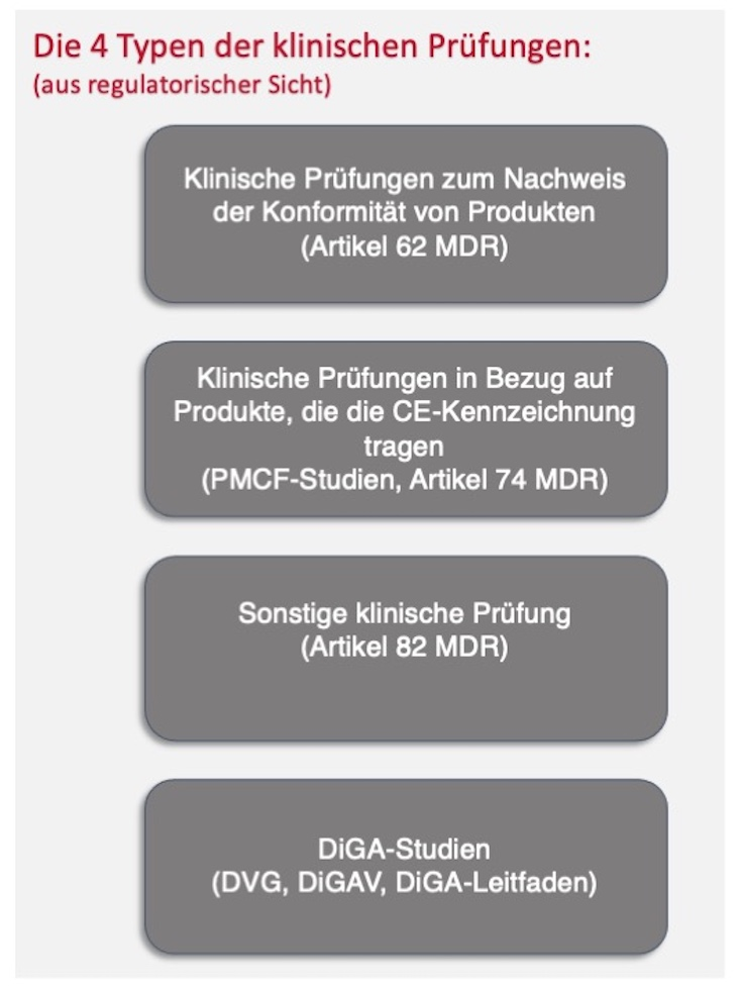
Abb. 1: Typen klinischer Prüfungen
Diese verschiedenen Typen unterscheiden sich hinsichtlich der jeweiligen regulatorischen Anforderungen und somit hinsichtlich der verschiedenen Schnittstellen zur technischen Dokumentation des zu untersuchenden Medizinproduktes.
Besonderheit bei PMCF-Studien:
Eine PMCF-Studie, die mit einem CE-gekennzeichneten Medizinprodukt im Rahmen der Zweckbestimmung und ohne belastende Untersuchungen stattfindet, stellt eine Ausnahme vom Artikel 74 der MDR dar:
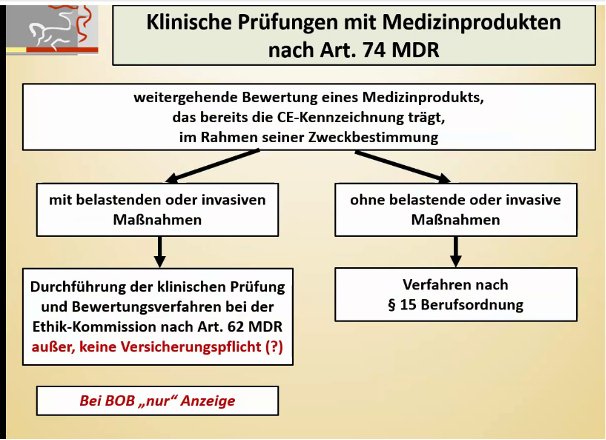
Abbildung 4: Artikel 74 der MDR (Quelle: Präsentationsfolien, BfArM-Veranstaltung, https://www.bfarm.de/DE/Service/Veranstaltungen/Dialogveranstaltungen/2021/210505-klinische_Pruefungen_von_MP.html)
Unabhängig davon sind aber dennoch die Dokumente nach Anhang XV Kapitel II und die Artikel 80ff der MDR verbindlich.
2. Dokumente, die bei PMCF-Studien einzureichen sind
Die folgenden Dokumente sind bei klinischen Prüfungen, die mit einem CE-gekennzeichneten Produkt zu erstellen. Abhängig davon, ob Artikel 74 der MDR zutrifft, erfolgt eine Einreichung bei der EK zur Votierung und bei BfArM. Gilt Artikel 74 nicht, ist lediglich eine berufsrechtliche Beratung nach § 15 der Berufsordnung für Ärzte (BO) bei der zuständigen Ethikkommission erforderlich. Egal, welche Antragsstellung erfolgt, simnd die folgenden Dokumente zu erstellen und diese haben einen Bezug zur technischen Dokumentation:
- Prüfplan - Anlagen gemäß Anhang XV Kap. II 3 MDR.
- Handbuch klinischer Prüfer -Anlagen gemäß Anhang XV Kap. II 2 MDR.
Folgende Dokumente werden hierzu aus der technischen Dokumentation benötigt:
- klinische Bewertung - gemäß Artikel 61 der MDR.
- Gebrauchsanweisung
- Ergebnisse biologische Sicherheitsprüfung
- Funktionsweise MP/Informationen zum MP (Funktionsweise MP)
- Funktionsweise und weitere Informationen zum Medizinprodukt.
- Risikoanalyse und -bewertung einschl. Restrisiken
- Liste grundlegende Sicherheits- und Leistungsanforderungen
- ggf. geeignete Aufbereitungs- oder Sterilisationsverfahren
- Nachweis CE-Kennzeichnung (Erforderlich, wenn das Prüfprodukt eine CE-Kennzeichnung trägt.)
2.1 Schnittstellen zur technischen Dokumentation
In der folgenden Tabelle sind die o. g. einzureichenden Dokumente sowie die darin enthaltenen Elemente und die jeweilige Entsprechung in der technischen Dokumentation aufgeführt:
|
Dokument |
Regulatorische Anforderung |
Elemente |
Technische Dokumentation |
|
Prüfplan |
Anhang XV Kap. II 3 MDR |
Kennzeichnung und Beschreibung des Produkts, einschließlich der Zweckbestimmung, des Herstellers, der Rückverfolgbarkeit, der Zielgruppe, der mit dem menschlichen Körper in Berührung kommenden Materialien, der mit seiner Verwendung verbundenen medizinischen und chirurgischen Verfahren und der für seine Verwendung erforderlichen Schulung und Erfahrung, der Sichtung der Referenzliteratur, des gegenwärtigen Stands der Technik bei der klinischen Versorgung in dem betreffenden |
Produktbeschreibung, Zweckbestimmung, Produktspezifikation, präklinischen Bewertung als Vorstufe der finalen klinischen Bewertung mit State of the Art Kapitel, Gebrauchsanweisung mit Beschreibung der Anwendung |
|
Prüfplan |
Anhang XV Kap. II 3 MDR |
Risiken und klinischer Nutzen des zu prüfenden Produkts |
Risikoanalyse, Risikomanagementbericht, präklinischen Bewertung mit Risiko-Nutzen-Abwägung |
|
Prüfplan |
Anhang XV Kap. II 3 MDR |
Informationen zu dem Prüfprodukt, zu etwaigen Komparatoren und anderen Produkten |
Produktbeschreibung, Gebrauchsanweisung |
|
Prüfplan |
Anhang XV Kap. II 3 MDR |
technische und funktionale Merkmale des Produkts |
Produktbeschreibung, Gebrauchsanweisung, Produktspezifikation |
|
Handbuch klinischer Prüfer |
Anhang XV Kap. II 2 MDR |
Kennzeichnung und Beschreibung des Produkts, einschließlich Informationen zur Zweckbestimmung, Risikoklassifizierung und geltenden Klassifizierungsregel gemäß Anhang VIII, Konzeption und Herstellung des Produkts sowie Verweis auf frühere und ähnliche Generationen des Produkts. |
Produktbeschreibung, Zweckbestimmung, Produktspezifikation, präklinischen Bewertung als Vorstufe der finalen klinischen Bewertung mit State of the Art Kapitel, Gebrauchsanweisung mit Beschreibung der Anwendung Klassifizierung |
|
Handbuch klinischer Prüfer |
Anhang XV Kap. II 2 MDR |
Herstellerangaben zur Installation, Wartung, Einhaltung von Hygienenormen und Verwendung, einschließlich Lagerungs- und Handhabungsbestimmungen, und — soweit diese Informationen vorliegen — die auf der Kennzeichnung anzubringenden Informationen und die Gebrauchsanweisung, die zusammen mit dem Produkt beim Inverkehrbringen bereitzustellen ist. |
-- |
|
Handbuch klinischer Prüfer/Präklinische Bewertung |
Anhang XV Kap. II 2.3 MDR |
Vorklinische Bewertung auf der Grundlage von Daten aus einschlägigen vorklinischen Tests und Versuchen, insbesondere aus Konstruktionsberechnungen, In-vitro-Tests, Ex-vivo-Tests, Tierversuchen, mechanischen oder elektrotechnischen Prüfungen, Zuverlässigkeitsprüfungen, Sterilisationsvalidierungen, Software-Verifizierungen und Validierungen, Leistungsversuchen, Bewertungen der Biokompatibilität und biologischen Sicherheit, sofern zutreffend. |
Präklinische Bewertung als Vorstufe der finalen klinischen Bewertung |
|
Handbuch klinischer Prüfer/Präklinische Bewertung |
Anhang XV Kap. II 2.3 MDR |
Bereits vorliegende klinische Daten, insbesondere — aus der einschlägigen verfügbaren wissenschaftlichen Fachliteratur zu Sicherheit, Leistung, klinischem Nutzen für die Patienten, Auslegungsmerkmalen und Zweckbestimmung des Produkts und/oder gleichartiger oder ähnlicher Produkte, — sonstige einschlägige verfügbare klinische Daten zu Sicherheit, Leistung, klinischem Nutzen für die Patienten, Auslegungsmerkmalen und Zweckbestimmung gleichartiger oder ähnlicher Produkte desselben Herstellers, einschließlich der Verweildauer des Produkts auf dem Markt, sowie die Daten aus einer Überprüfung der Leistungs- und Sicherheitsaspekte und des klinischen Nutzens und etwaigen unternommenen Korrekturmaßnahmen. |
Präklinische Bewertung als Vorstufe der finalen klinischen Bewertung |
|
Handbuch klinischer Prüfer/Präklinische Bewertung |
Anhang XV Kap. II 2.3 MDR |
Zusammenfassung der Nutzen-Risiko-Analyse und des Risikomanagements, einschließlich Informationen zu bekannten oder vorhersehbaren Risiken, etwaigen unerwünschten Nebenwirkungen, Kontraindikationen und Warnhinweisen. |
Gebrauchsanweisung, präklinische Bewertung als Vorstufe der finalen klinischen Bewertung |
|
Handbuch klinischer Prüfer |
Anhang XV Kap. II 2 MDR |
Bei Produkten, zu deren Bestandteilen ein Arzneimittel, einschließlich eines Derivats aus menschlichem Blut oder Plasma gehört oder bei Produkten, die unter Verwendung nicht lebensfähiger Gewebe oder Zellen menschlichen oder tierischen Ursprungs oder ihren Derivaten hergestellt werden. |
Nur in diesem Fall: Informationen zu dem Arzneimittel bzw. den Geweben, den Zellen oder ihren Derivaten sowie zur Erfüllung der relevanten grundlegenden Sicherheits- und Leistungsanforderungen und dem spezifischen Risikomanagement bezüglich des Arzneimittels bzw. der Gewebe oder Zellen oder ihrer Derivate sowie einen Nachweis des durch die Einbeziehung dieser Bestandteile bezüglich des klinischen Nutzens und/oder der Sicherheit des Produkts entstehenden Mehrwerts |
|
Handbuch klinischer Prüfer |
Anhang XV Kap. II 2 MDR |
Ein Verzeichnis, aus dem die Erfüllung der einschlägigen grundlegenden Sicherheits- und Leistungsanforderungen des Anhangs I einschließlich der — vollständig oder in Teilen — angewandten Normen und Spezifikationen im Einzelnen hervorgeht, sowie eine Beschreibung der zur Erfüllung der einschlägigen grundlegenden Sicherheits- und Leistungsanforderungen gewählten Lösungen, soweit diese Normen und Spezifikationen nur zum Teil oder überhaupt nicht erfüllt sind oder gänzlich fehlen. |
Checkliste der grundlegenden Leistungs- und Sicherheitsanforderungen Normenliste |
|
Gebrauchsanweisung |
Anhang XV Kap. II 2.2 MDR |
-- |
Gebrauchsanweisung |
|
Versicherung grundlegende Sicherheits- und Leistungsanforderungen |
Anhang XV Kap II 4.1 MDR |
Versicherung, dass die grundlegenden Sicherheits- und Leistungsanforderungen erfüllt sind. |
Checkliste der grundlegenden Leistungs- und Sicherheitsanforderungen |
|
Ergebnisse biologische Sicherheitsprüfung |
Anhang XV Kap. II 2.3 MDR |
Daten aus einschlägigen vorklinischen Tests und Versuchen, insbesondere Bewertungen der Biokompatibilität und biologischen Sicherheit, sofern zutreffend. |
Testberichte, Bericht zur biologischen Sicherheit |
|
Funktionsweise MP/Informationen zum MP |
-- |
Funktionsweise und weitere Informationen zum Medizinprodukt |
Produktbeschreibung, Gebrauchsanweisung, Produktspezifikation |
|
Risikoanalyse und -bewertung einschl. Restrisiken |
Anhang XV Kap. II 2.5 bzw. 4.6 MDR |
Zusammenfassung der Nutzen-Risiko-Analyse und des Risikomanagements, einschließlich Informationen zu bekannten oder vorhersehbaren Risiken, etwaigen unerwünschten Nebenwirkungen, Kontraindikationen und Warnhinweisen. Der zuständigen Behörde, die einen Antrag überprüft, werden vollständige Angaben zu der verfügbaren technischen Dokumentation, zum Beispiel detaillierte Unterlagen zu Risikoanalyse/-management oder spezifische Testberichte, auf Anfrage vorgelegt. |
Risikomanagementdokumentation gemäß ISO 14971 |
|
Liste grundlegende Sicherheits- und Leistungsanforderungen |
Anhang XV Kap. II 2.7 MDR |
-- |
Checkliste der grundlegenden Leistungs- und Sicherheitsanforderungen |
|
ggf. geeignete Aufbereitungs- oder Sterilisationsverfahren |
-- |
Sterilisationsprozess, Validierung |
Dokumentation zum Sterilisationsverfahren |
Tabelle 1: Dokumente der klinischen Prüfung und technische Dokumentation
Da es sich um ein CE-gekennzeichnetes Produkt handelt, sind all diese Dokumente bereits in der technischen Dokumentation vorhanden und fließen insbesondere in den Prüfplan sowie in das Handbuch des klinischen Prüfers mit ein.
Das CE-gekennzeichnetes Medizinprodukt ist damit das in der PMCF-Studie geprüfte Prüfprodukt, für das die klinischen Daten erhoben werden. D. h. die Dokumente und Ergebnisse müssen sich auf genau dieses Produkt und nicht auf eine frühere Version beziehen!
Technische Dokumentation:
- Produktbeschreibung
- Zweckbestimmung
- Produktspezifikation
- finale klinischen Bewertung mit State of the Art Kapitel und Literatur und Risiko-Nutzen-Abwägung
- Risikomanagementdokumentation gemäß ISO 14971: PHA, Risikoanalyse, Risikomanagementbericht
- Gebrauchsanweisung
- Klassifizierung
- Checkliste der grundlegenden Leistungs- und Sicherheitsanforderungen
- Normenliste
- Verifizierungstestberichte
- Bericht zur biologischen Sicherheit (falls zutreffend)
- Dokumentation zum Sterilisationsverfahren (falls zutreffend)
2.2 Synergien
Schaut man sich die oben aufgeführte Liste an, fällt auf, dass es sich hierbei bereits nahezu um die gesamte technische Dokumentation handelt.
Ein gutes Beispiel für die Nutzung der Synergien bei der Erstellung der Dokumente ist der Prüfplan. Sehr viele Abschnitt sind dieselben Inhalte wie bereits in anderen Dokumenten der technischen Dokumentation. Die Zweckbestimmung, die Produktbeschreibung usw. sind nur wenige Beispiele. Außerdem werden in der finalen klinischen Bewertung klinische Daten zum Stand der Technik aufgeführt, die ebenfalls für den Prüfplan und das Handbuch des klinischen Prüfers verwendet werden können.
Und hier spielt erneut die Digitalisierung der klinischen Prüfung eine Rolle:
Mit der engen Verzahnung der klinischen Prüfung nicht nur mit dem Prozess der Literatursuche und somit mit der klinischen Bewertung wie im vorletzten Blogbeitrag berichtet, sondern auch mit der technischen Dokumentation ist auch eine Digitalisierung der wesentlichen Dokumente der klinischen Prüfung wie z. B.
- Klinischer Prüfplan (Anhang XV, Kapitel II, Abschnitt 3 der MDR)
- Handbuch des klinischen Prüfers (Anhang XV, Kapitel II, Abschnitt 2 der MDR)
- klinische Bewertung
möglich.
Die Vorteile der Digitalisierung liegen auf der Hand:
- effizienteres Arbeiten
- zielorientiertes Einsetzen der Kapazitäten
- Beseitigung von Ineffizienzen bei Erstellung, Pflege und Änderung von Inhalten der Technischen Dokumentation, klinischen Bewertung und Literatursuchen
- langfristige Verringerung des Pflegeaufwands
Über die Softwareapplikation "Polarion" lassen sich Schnittstellen wie Zweckbestimmung, Risikomanagement, Gebrauchstauglichkeit, klinische Bewertung, klinische Prüfung Projekten zuordnen und bei Bedarf wiederverwenden. Die Erstellung und Pflege von Dokumenten wird somit deutlich vereinfacht und beschleunigt. Daneben werden Redundanzen und Inkonsistenzen vermieden.
3. Ausblick
Unser „Adventsspezial“ beschäftigt sich nächste Woche mit den Schnittstellen zur technischen Dokumentation bei DiGA-Studien.
4. Wie wir Ihnen helfen können
Ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss, klären wir bei medXteam im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung.
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung