Die Blog-Reihe zu den Typen klinischer Prüfungen wird im Dezember durch unser „Weihnachtsspezial“ unterbrochen. Hiermit möchten wir Sie umfassend über die wichtigen Änderungen in Bezug auf klinische Prüfungen durch die MDR noch dieses Jahr informieren, damit Sie für 2021 gewappnet sind.
Das Besondere an unserer Aktion ist dabei, dass der Beitrag bis Weihnachten wächst. Jede Woche kommen neue Abschnitte dazu. Im Januar wird es dann mit dem Thema DiGA-Studien weitergehen.
Der erste Teil unseres Dezemberspecials gab Ihnen einen Leitfaden für das Antragsverfahren für klinische Prüfungen im Rahmen des Konformitätsbewertungsverfahrens bei der Bundesoberbehörde und der Ethikkommissionen. Der heutige zweite Teil beschäftigt sich nun mit dem Antragsverfahren für klinische Prüfungen mit CE-gekennzeichneten Produkten.
Abkürzungen.
BOB (Bundesoberbehörde)
EK (Ethikkommission)
KP (klinische Prüfung)
MDR (medical device regulation; Verordnung 2017/745)
MPEUAnpG (das Medizinprodukte-EU-Anpassungsgesetz wurde am 25.05.2020 vom Bundestag als Gesetze verabschiedet. Dieses MPAnpG-EU beschreibt im Artikel 1 das Medizinprodukte-Durchführungsgesetz (MPDG))
MPDG (das MPDG wird das Medizinproduktegesetz (MPG) ab 26. Mai 2021 schrittweise ablösen und für alle Hersteller und Betreiber von Medizinprodukten in Deutschland rechtsverbindlich sein).
Teil 2: Antragsverfahren – Genehmigungsprozess für klinische Prüfungen mit CE-gekennzeichneten Produkten - Artikel 74 MDR
1. Einleitung
Aktuell noch und bis zur Gültigkeit der MDR ab 21. Mai 2021 gelten für die Genehmigung klinischer Prüfungen aktuell die §§ 20 ff MPG für klinische Prüfungen, die in Deutschland durchgeführt werden. Für klinische Prüfungen mit CE-gekennzeichneten Produkten findet sich die "Ausnahme" zu klinischen Prüfungen nach § 20 MPG im § 23b MPG. Dieser deckt die sog. PMCF-Studien ab. Hierfür gilt außerdem die dem MPG untergeordnete MEDDEV Guideline für Post-Market Clinical Follow-up Studies (MEDDEV 2.12/2 Rev. 2). Das bedeutet, dass hier lediglich ein Antrag auf berufsrechtliche Beratung bei der für das Prüfzentrum und den Prüfer zuständigen Ethikkommission nach § 15 BO (Berufsordnung der Ärzte) zu stellen ist und die Ethikkommission (EK) kein Votum abgibt. Das gilt allerdings nur, wenn das Produkt im Rahmen dieser klinischen Prüfung im Rahmen seiner Zweckbestimmung angewendet wird und/oder keine zusätzlichen belastenden Untersuchungen stattfinden. Ist dies der Fall, greift wieder § 20 MPG. Das zeigen auch die beiden folgenden Abbildungen:
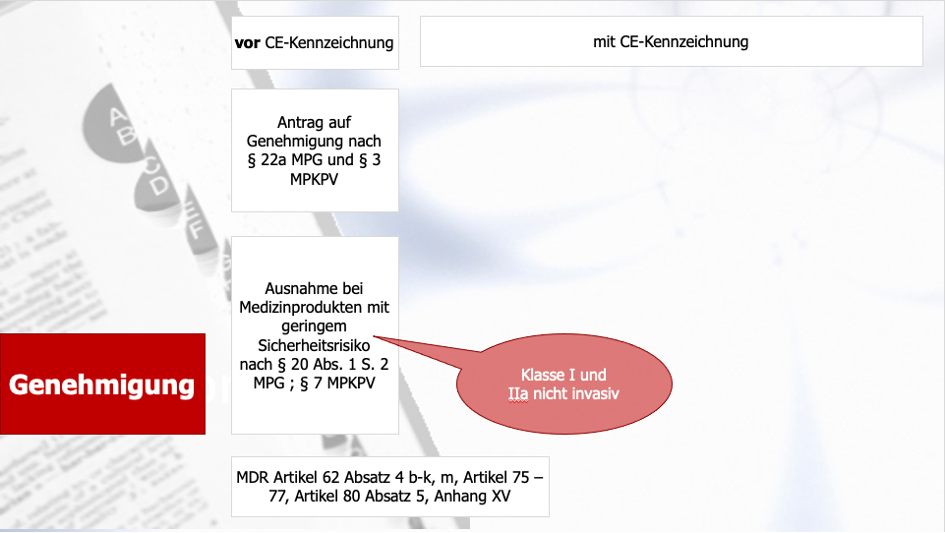
Abbildung 1: Bisheriger Genehmigungsprozess vor der CE-Kennzeichnung (Änderungen durch die MDR - Artikel)
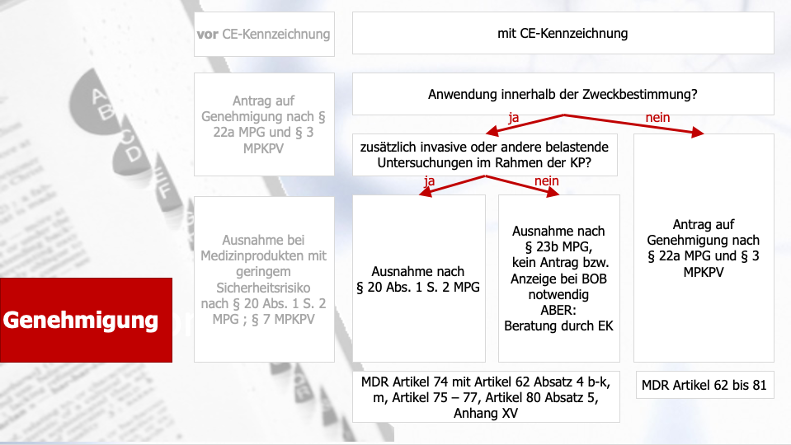
Abbildung 2: Bisheriger Genehmigungsprozess nach der CE-Kennzeichnung (Änderungen durch die MDR - Artikel)
Das alles ändert sich nun mit der MDR, da dort das Genehmigungsverfahren und im zugehörigen MPDG das nationale Verfahren für die EKs beschrieben und festgelegt ist. In beiden Abbildungen oben sind dazu schon die entsprechenden Artikel der MDR aufgeführt. So verschwindet z. B. der § 23b MPG völlig. Das ist mit eine der größten Veränderungen und Auswirkungen durch die MDR, da die berufsrechtliche Beratung durch die Ethikkommission nun entfällt. Was hier zukünftig ab Mai 2021 zu beachten ist, wird nun im Folgenden beschrieben.
2. Genehmigungsprozess für klinische Prüfungen mit CE-gekennzeichneten Produkten - Artikel 74 MDR
Für die sogenannten „PMCF-Studien“ (Post-Market-Follow-up = klinische Nachbeobachtung nach dem Inverkehrbringen des Produktes) wird unterschieden, ob das Prüfprodukt innerhalb der Zweckbestimmung angewendet wird oder nicht. Bisher wurde hier der o. g. § 23b MPG und der § 7 der MPKPV angewendet. Beides wurde nicht ins MPAnpG übernommen (hier wird nur das nationale Antragsverfahren bei der EK geregelt), sondern in der MDR abgedeckt:
2.1 Das Medizinprodukt mit CE-Zeichen wird im Rahmen seiner Zweckbestimmung angewendet
Folgende Artikel der MDR kommen zur Anwendung:
- Artikel 74 Absatz 1 und
- dann gelten die Regeln des § 62 Absatz 4, Buchstaben b bis k und m,
- Artikel 75 bis 77 Artikel 80 Absatz 5 (Vigilanz) sowie
- die Bestimmungen des Anhangs XV der MDR und hier die geforderten Dokumente aus Kapitel II
Was bedeutet das?
Es wird eine Stellungnahme von der EK wie bei einer Zulassungsstudie (s. Teil 1 des Weihnachtsspezials) gefordert. Das bedeutet, mit der Gültigkeit der MDR wird auch für PMCF-Studien ein EK-Votum benötigt. (Artikel 62 Absatz 4 Satz b)
Ein Antrag bei der BOB muss weiterhin unter diesen Voraussetzungen nicht gestellt werden.
Achtung: Sollten im Rahmen der PCMF Studie zusätzliche invasive oder belastende Verfahren angewendet werden, so muss der Sponsor mindestens 30 Tage vor Beginn der klinischen Prüfung die BOB informieren und die Unterlagen gemäß Anhang XV Kapitel II übermitteln. Bisher wurde das über den § 23b MPG in Verbindung mit dem § 7 der MPKPV geregelt und BfArM führte lediglich eine weniger umfangreiche Überprüfung insbesondere im Hinblick auf die Sicherheitsaspekte der zusätzlichen belastenden Untersuchung durch.
Hier wird dann wie oben ein EK-Votum benötigt aber die Unterlagen müssen außerdem über EUDAMED bei BfArM eingereicht werden
Was bedeutet das?
Ein Votum der EK wird wie oben weiterhin benötigt und zusätzlich dazu muss nun ein Antrag auch bei BfArM über EUDAMED gestellt werden.
Die Antragsfristen sind diesbezüglich dieselben wie bei den klinischen Prüfungen im Rahmen des Konformitätsbewertungsverfahrens (s. Teil 1).
Welche Auswirkungen hat das?
Die Änderung hin zum EK-Votum bei PMCF-Studien bedeutet eine vollumfängliche Prüfung auch der Qualifikation von Hauptprüfer und Prüfer: Die MDR sagt in Artikel 62 Absatz 4 Satz j dazu:
die Verantwortung für die medizinische Versorgung der Prüfungsteilnehmer trägt ein Arzt mit geeigneter Qualifikation oder gegebenenfalls ein qualifizierter Zahnarzt oder jede andere Person, die nach nationalem Recht zur Bereitstellung der entsprechenden Patientenbetreuung im Rahmen einer klinischen Prüfung befugt ist...
Und das wird national im § 30 des MPAnpG noch detailliert:
(4) Als Leiter einer klinischen Prüfung oder einer sonstigen klinischen Prüfung kann nur bestimmt werden, wer eine mindestens zweijährige Erfahrung in der klinischen Prüfung von Medizinprodukten nachweisen kann.
(5) Der Nachweis der [...] geforderten Qualifikation ist durch einen aktuellen Lebenslauf und durch andere aussagefähige Dokumente zu erbringen.
Das bedeutet, dass zukünftig auch bei PMCF-Studien als Qualifikationsvoraussetzung eine zweijährige Erfahrung mit Medizinproduktestudien gefordert wird. Da dies im Rahmen der berufsrechtlichen Beratung der EK nach § 23b MPG und § 15 BO nicht gefordert war, stellt dies ab Mai 2021 eine größere Hürde auch für die Durchführung von PMCF-Studien dar.
2.2 Das Medizinprodukt mit CE-Zeichen wird außerhalb seiner Zweckbestimmung angewendet
Wird das CE-gekennzeichnete Produkt im Rahmen der Studie außerhalb seiner Zweckbestimmung angewendet (z. B. um klinische Daten für eine neue Indikation zu erheben), so gelten die Artikel 62 bis 81 wie bei einer Zulassungsstudie auch. Bisher wurde das ebenfalls über den § 23b MPG in Verbindung mit dem § 7 der MPKPV geregelt und BfArM führte lediglich eine weniger umfangreiche Überprüfung insbesondere im Hinblick auf die Sicherheitsaspekte der erweiterten Zweckbestimmung durch. Im Rahmen der MDR ändert sich dies nun und es gelten somit:
- Artikel 62 bis 81, d. h. wie bei einer Zulassungsstudie (Teil 1 des Weihnachtsspezials)
- Anhang XV und hier Dokumente aus Kapitel II
Es wird eine Stellungnahme von der EK wie bei einer Zulassungsstudie (s. Teil 1 des Weihnachtsspezials) gefordert. Außerdem muss wie bei Zulassungsstudien ein Antrag bei BfArM (über EUDAMED) gestellt werden.
Die Antragsfristen sind auch hier dieselben wie bei den klinischen Prüfungen im Rahmen des Konformitätsbewertungsverfahrens (s. Teil 1).
3. Ausblick
Das war nun Teil 2 unseres „Weihnachtsspezial“ und nächste Woche geht es dann weiter mit dem Genehmigungsverfahren bei sonstigen klinischen Prüfungen. Auch damit werden wir Sie wieder umfassend über die wichtigen Änderungen in Bezug auf klinische Prüfungen durch die MDR noch dieses Jahr informieren, damit Sie für 2021 gewappnet sind.
Das Besondere an unserer Aktion ist dabei, dass der Beitrag bis Weihnachten wächst. Jede Woche kommen neue Abschnitte mit weiteren Änderungen dazu.
Im Januar wird es dann mit dem Thema DiGA-Studien weitergehen.
4. Wie wir Ihnen helfen können
Ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss, klären wir bei medXteam im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung.
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung