Bei medXteam stehen klinische Daten im Mittelpunkt. In diesem Kontext führen wir als CRO nicht nur klinische Prüfungen mit Medizinprodukten gemäß MDR und ISO 14155 durch, sondern bieten auch sämtliche weiteren Möglichkeiten und Formen der Datenerhebung an. Egal, für welche Form der Datenerhebung man sich entscheidet: Das Fundament bildet eine solide Planung aber auch das Auseinandersetzen mit den verschiedenen Optionen und den jeweiligen Anforderungen daran. Ein schönes Beispiel dafür, dass mit der MDR Anforderungen nicht nur stringenter werden und zunehmen ist das, was in diesem Blog-Beitrag dargestellt wird: Nämlich die geänderte Qualifikationsanforderung an das Studienpersonal und die Konsequenzen, die daraus resultieren, die in bestimmten Fällen sogar zu einer Erleichterung führen.
Abkürzungen
MDR Medical Device Regulation; EU-Verordnung 2017/745
MPDG Medizinproduktedurchführungsgesetz
MPAnpG Medizinprodukteanpassungsgesetz
MPG Medizinproduktegesetz
LKP Leiter der klinischen Prüfung
Zugrundeliegende Regularien
EU-Verordnung 2017/745 (MDR)
Medizinprodukte-Durchführungsgesetz (MPDG)
1. Einleitung
Die rasante Entwicklung in der Medizintechnikbranche zieht eine stetige Anpassung und Weiterentwicklung der gesetzlichen Rahmenbedingungen nach sich. Insbesondere die Einführung der EU-Verordnung 2017/745, besser bekannt als Medical Device Regulation (MDR), und die daraus in Deutschland abgeleitete nationale Gesetzgebung durch das Medizinprodukte-Anpassungsgesetz (MPAnpG) und das Medizinproduktedurchführungsgesetz (MPDG) haben tiefgreifende Auswirkungen auf die Planung und Durchführung von klinischen Prüfungen.
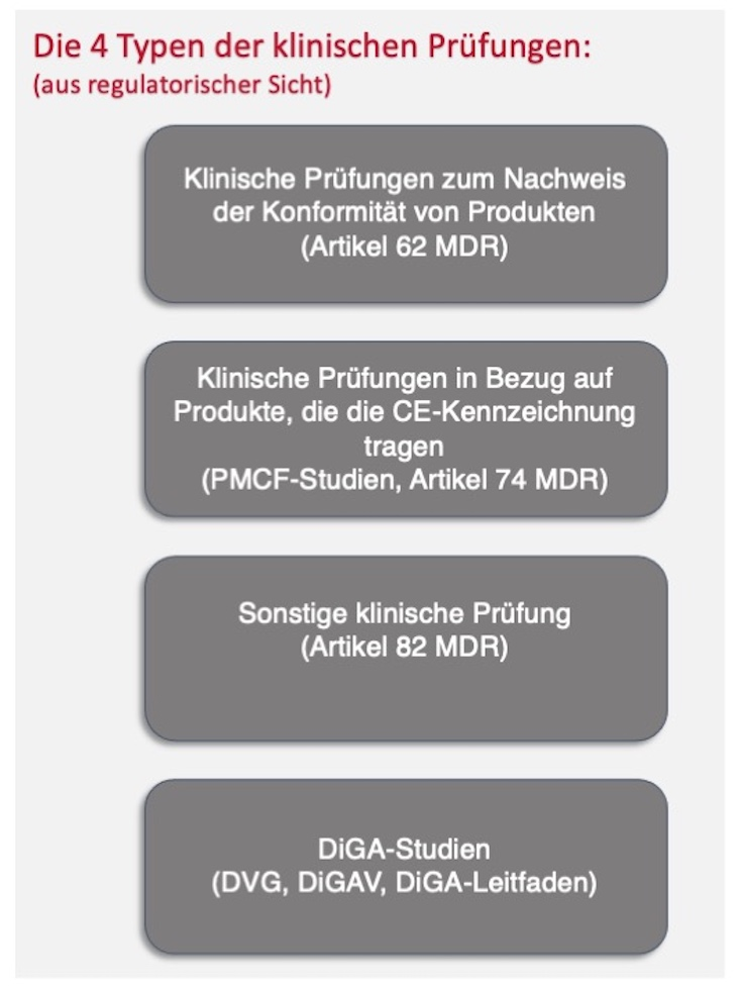
Die Überlegung, ob man sich bei klinischen Prüfungen nach Artikel 62, 74 oder 82 der MDR für ein monozentrisches oder ein multizentrisches Design entscheidet, spielt dabei eine zentrale Rolle. Denn obwohl die MDR und das MPDG in vielen Bereichen eine strengere Regulierung mit sich bringen, gibt es in bestimmten Aspekten auch deutliche Erleichterungen. Eine solche Erleichterung betrifft insbesondere das Design von klinischen Prüfungen. So sind beispielsweise bei monozentrischen Studien die Genehmigungshürden deutlich niedriger. Aber welche genauen Vorteile bieten sie, und welche Herausforderungen und Anforderungen ergeben sich daraus an insbesondere die Qualifikation des erforderlichen Studienpersonals?
Dieser Blogbeitrag bringt Licht ins Dunkel und hebt die entscheidenden Unterschiede sowie die damit verbundenen regulatorischen und organisatorischen Überlegungen im Kontext des Designs klinischer Prüfungen hervor. Dabei wird auch ein besonderes Augenmerk auf die Rolle und die Anforderungen des Studienpersonals gelegt, die durch die neue Gesetzgebung deutlich definiert und in den Mittelpunkt gerückt werden.
2. Monozentrische Studie vs. multizentrische Studie
Das Design einer klinischen Prüfung hängt von verschiedenen Faktoren ab, einschließlich der Art des Medizinprodukts, der Zielsetzung der Studie und den verfügbaren Ressourcen. Je nachdem, welches Design gewählt wird, ergeben sich unterschiedliche Anforderungen an das Studienpersonal und die Organisation der Studie.
Die Wahl des Designs für klinische Prüfungen, ob monozentrisch oder multizentrisch, hat tiefgreifende Auswirkungen auf die Umsetzung, das Budget, die Zeitplanung und die Datenqualität.
2.1 Monozentrische Studie
Bei einer monozentrischen Studie handelt es sich um eine klinische Prüfung, die in einem einzigen Zentrum oder an einem einzigen Ort durchgeführt wird. Das Studienteam besteht in der Regel aus einem Prüfer. Bei größeren Studien an einem Standort oder wenn verschiedene Fachbereiche involviert sind, kann das Team jedoch auch aus mehreren Prüfern bestehen. In diesem Fall wird einer der Prüfer als Hauptprüfer eingesetzt, der für die Gesamtkoordination der Studie verantwortlich ist. Zusätzlich können weitere Beteiligte wie Study Nurses, die für die Patientenbetreuung und Datenerfassung verantwortlich sind, zum Studienteam gehören.
Vorteile von monozentrischen Studien:
- Einfachheit: Da nur ein Standort involviert ist, sind die Prozesse in der Regel weniger komplex.
- Kosten: Da weniger Personal und Ressourcen benötigt werden, sind die Kosten in der Regel niedriger.
- Kontrolle: Der Prüfer oder Hauptprüfer hat eine direkte Übersicht und Kontrolle über alle Aspekte der Studie.
- Schnellere Kommunikation: Mit einem kleineren Team und nur einem Standort sind Absprachen und Entscheidungsprozesse in der Regel schneller und direkter.
Diese Einfachheit und Kostenersparnis können jedoch durch den begrenzten Patientenpool und die geografische Begrenzung kompensiert werden. Es besteht das Risiko, dass die Ergebnisse nicht allgemeingültig sind oder dass es schwierig ist, genügend Patienten für die Studie zu rekrutieren. Wenn viele Patienten gemäß statistischer Fallzahlplanung benötigt werden, kann diese Form des Designs nicht gewählt werden, da dies dann nicht in einem angemessenen Zeitrahmen umsetzbar ist.
2.2 Multizentrische Studie
Multizentrische Studien sind klinische Prüfungen, die an mehreren Standorten oder Zentren durchgeführt werden. Bei solchen Studien besteht das Studienteam in jedem Zentrum typischerweise aus einem Prüfer, einer Study Nurse und gegebenenfalls weiteren beteiligten Fachkräften. Trotz der Mehrfachzentrenstruktur bleibt in jedem Zentrum der Ablauf ähnlich wie bei monozentrischen Studien. Der Unterschied besteht darin, dass ein derartiges Studiendesign ein Hauptprüfzentrum erfordert. Dieses Hauptprüfzentrum stellt den Leiter der klinischen Prüfung (LKP), der die gesamte Studie über alle Zentren hinweg koordiniert.
Vorteile von multizentrischen Studien:
- Patientenpool: Durch die Beteiligung mehrerer Zentren wird der Zugang zu einer größeren und heterogen Patientenpopulation ermöglicht.
- Datenbasis: Das Design ermöglicht eine breitere und repräsentativere Datenerhebung, da sie aus verschiedenen Populationen und Standorten stammt. Die wissenschaftliche Aussagekraft erhöht sich zudem durch die Involviertheit mehrerer Prüfer, die externe Validität wächst.
- Vergleichbarkeit: Durch verschiedene Standorte können direkte Vergleiche und Konsistenzüberprüfungen durchgeführt werden.
Jedoch können diese Vorteile durch die erhöhten Kosten, den größeren organisatorischen Aufwand und die Koordinationsanforderungen zwischen den Zentren ausgeglichen werden.
- Kernfaktoren: Kosten, Aufwand und Studienpersonal:
- Kosten: Multizentrische Studien können aufgrund ihrer Größe und Komplexität teurer sein als monozentrische Studien.
- Aufwand: Der organisatorische Aufwand für multizentrische Studien ist bedeutend höher, insbesondere in Bezug auf die Koordination von Patientenrekrutierung, Datenmanagement und Kommunikation zwischen den Zentren.
Studienpersonal: Hier liegt einer der kritischsten Aspekte. Die Herausforderung besteht darin, konsistente Protokolle und Praktiken über alle Zentren hinweg sicherzustellen. Dieser besondere Knackpunkt und die damit verbundenen Überlegungen und Strategien werden im Folgenden detailliert behandelt.
3. Entwicklung der Qualifikationsanforderungen für Prüfer: Vom MPG zum MPAnpG
Mit der Fortentwicklung der regulatorischen Landschaft für Medizinprodukte in Deutschland haben sich auch die Qualifikationsanforderungen für Personen, die an der Durchführung klinischer Prüfungen beteiligt sind, gewandelt.
3.1 Unter dem Medizinproduktegesetz (MPG)
Gemäß § 20 des Medizinproduktegesetzes (MPG) mussten klinische Prüfungen bestimmte Voraussetzungen erfüllen. Ein entscheidender Aspekt war, dass sie in einer geeigneten Einrichtung durchgeführt und von einem "angemessen qualifizierten Prüfer" geleitet werden mussten. Das MPG gab klare Vorgaben zur Qualifikation dieses Prüfers: Neben der medizinischen oder zahnmedizinischen Ausbildung musste er mindestens eine zweijährige Erfahrung in der klinischen Prüfung von Medizinprodukten nachweisen können.
Diese Anforderung galt für alle Prüfer, egal ob Prüfer, Hauptprüfer oder LKP.
3.2 Übergang zum Medizinprodukte-Anpassungsgesetz (MPAnpG oder MDPG)
Mit der Einführung des Medizinprodukte-Anpassungsgesetzes wurden die Anforderungen an die Qualifikationen des Studienpersonals präzisiert und erweitert.
Nachdem im MPG keine Rollendefinition vorhanden war und diese vor dem Inkrafttreten des MPDG aus der ISO 14155 zu entnehmen war, definiert nun das MPDG zumindest die Rollen Hauptprüfer und LKP in § 3 (5,6):
"Nach § 30 des MDPG gibt es klare Unterscheidungen zwischen dem Prüfer, dem Hauptprüfer und dem Leiter einer klinischen Prüfung. Während der Hauptprüfer und Prüfer weiterhin wichtige Rollen in der klinischen Prüfung spielen, wird die besondere Qualifikation der mindestens zweijährigen Erfahrung in der klinischen Prüfung von Medizinprodukten nun explizit dem Leiter einer klinischen Prüfung oder einer sonstigen klinischen Prüfung zugewiesen."
Das bedeutet, dass sich im Vergleich zum vorherigen MPG die Qualifikationsanforderungen konkretisiert haben und spezifischer auf die unterschiedlichen Rollen im klinischen Prüfprozess abgestimmt sind. Dies zeigt ein gestiegenes Bewusstsein für die Notwendigkeit klar definierter und strenger Qualifikationskriterien, um die Qualität und Integrität von klinischen Prüfungen zu gewährleisten. Es spiegelt auch die wachsende Komplexität und Bedeutung klinischer Prüfungen im Prozess der Medizinprodukteentwicklung und -zulassung wider.
3.3 Konsequenzen und Auswirkungen
Die fortschreitende Anpassung und Verfeinerung der gesetzlichen Rahmenbedingungen hat einen erheblichen Einfluss darauf, wie klinische Prüfungen von Medizinprodukten durchgeführt werden. Insbesondere hat das Medizinproduktedurchführungsgesetz (MPDG) einige grundlegende Veränderungen hervorgebracht, die die Organisation und Genehmigung klinischer Prüfungen beeinflussen.
Eine solche bedeutsame Veränderung, die im MPDG vorgenommen wurde, betrifft somit die Rollen und Qualifikationen der Personen, die an klinischen Prüfungen beteiligt sind. Laut § 30 des MDPG wird nun differenziert zwischen dem Prüfer, dem Hauptprüfer und dem Leiter einer klinischen Prüfung. Es ist von entscheidender Bedeutung zu erkennen, dass die zuvor im MPG geforderte mindestens zweijährige Erfahrung in der klinischen Prüfung von Medizinprodukten eines jeglichen Prüfers auch schon bei einer monozentrischen Studie jetzt nur noch dem Leiter einer klinischen Prüfung oder einer sonstigen klinischen Prüfung explizit zugeordnet wird. Somit trifft diese Anforderung nur noch multizentrische Studien.
Prüfer und Hauptprüfer spielen zwar weiterhin wichtige Rollen im Prozess, doch die besondere Qualifikationsanforderung bezieht sich nun nur noch auf den Leiter der Prüfung.
Für monozentrische Studien bedeutet dies, dass der Genehmigungsprozess einer klinischen Prüfung erheblich vereinfacht wird. Durch die Fokussierung der zweijährigen Erfahrungsanforderung auf den Leiter einer klinischen Prüfung und nicht auf jeden beteiligten Prüfer, wird die Hürde für die Durchführung solcher Studien damit erheblich gesenkt, da die Ethikkommission diese Anforderung für den Prüfer nicht mehr erwartet und somit nicht mehr prüft.
4. Schlussfolgerung
Das Fazit aus diesen Beobachtungen ist evident: Die Wahl zwischen monozentrischem und multizentrischem Design hat erhebliche Auswirkungen auf die Genehmigung, Kosten, den organisatorischen Aufwand und die Anforderungen an das Studienpersonal einer klinischen Prüfung. Die richtige Planung und Berücksichtigung aller relevanten Aspekte sind daher unerlässlich für den Erfolg des Projekts. Es ist entscheidend, sich intensiv mit den Anforderungen auseinanderzusetzen und das geeignete Personal für die jeweilige Studienart auszuwählen.
Die Wahl zwischen monozentrischem und multizentrischem Design hat erhebliche Auswirkungen auf die Genehmigung, Kosten, den Aufwand und die Anforderungen an das Studienpersonal einer klinischen Prüfung. Die richtige Planung und Berücksichtigung aller relevanten Aspekte sind daher unerlässlich für den Erfolg des Projekts. Es ist entscheidend, sich intensiv mit den Anforderungen auseinanderzusetzen und das geeignete Personal für die jeweilige Studienart auszuwählen.
Gerade dieser Aspekt beeinflusst maßgeblich die Durchführung von klinischen Prüfungen. Das Medizinproduktedurchführungsgesetz (MPDG) hat hierbei signifikante Änderungen eingeführt, insbesondere in Bezug auf die Rollen und Qualifikationen der Beteiligten einer klinischen Prüfung.
Durch den § 30 des MDPG und die damit verbundene klare Unterscheidung zwischen dem Prüfer, dem Hauptprüfer und dem Leiter einer klinischen Prüfung, hat sich der Genehmigungsprozess für monozentrische Studien wesentlich vereinfacht. Die Spezifizierung, dass die mindestens zweijährige Erfahrung in der klinischen Prüfung von Medizinprodukten nun explizit dem Leiter einer klinischen Prüfung zugeschrieben wird, eröffnet neue Möglichkeiten in der Gestaltung klinischer Prüfungen und senkt die Hürden ihrer Durchführung.
Ein tiefergehendes Verständnis der gesetzlichen Anforderungen und eine sorgfältige Auswahl des geeigneten Personals für die spezifische Studienart sind dabei unabdingbare Schlüsselaspekte. Es ist von zentraler Bedeutung, sich intensiv mit diesen Anforderungen auseinanderzusetzen und die Planung entsprechend auszurichten.
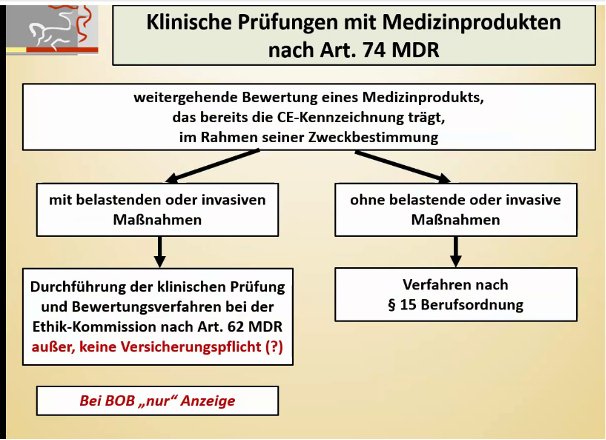
Dies bezieht sich allerdings ausschließlich auf klinische Prüfungen, die im Rahmen der MDR durchgeführt werden (Artikel 62, 74 und 82). Bei allen anderen klinischen Prüfungen (z. B. PMCF-Studien innerhalb der Zweckbestimmung des Medizinprodukts und ohne belastende Untersuchungen bleiben hiervon unberührt. Das heißt, hier gibt es bei multizentrischen Studien keine Anforderungen dieser Art an den LKP.
5. Was wir für Sie tun können
Wenn eine klinische Prüfung durchgeführt werden soll, müssen zuvor grundlegende Sicherheits- und Leistungsanforderungen erfüllt und somit wesentliche Dokumente der Technischen Dokumentation erstellt werden.
Außerdem benötigen alle Hersteller von Medizinprodukten ein QMS, auch bei der Entwicklung von Produkten der Klasse I.
Die klinische Prüfung mündet in der klinischen Bewertung, die dann wiederum die Basis für PMCF-Aktivitäten (einschließlich einer PMCF-Studie) darstellt.
Wir unterstützen Sie deshalb während Ihres kompletten Vorhabens mit Ihrem Medizinprodukt immer mit primärem Bezug auf die klinischen Daten zum Produkt: von Anfang an bis zum Ende.
6. Wie wir Ihnen helfen können
Ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss, klären wir bei medXteam im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung.
Wir unterstützen außerdem im Bereich der Entwicklungsstrategie, der technischen Dokumentation und im Rahmen des Qualitätsmanagements.
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung