Bei medXteam stehen klinische Daten im Mittelpunkt. Wir erheben diese über Literatursuchen oder direkt mit dem Medizinprodukt im Rahmen klinischer Prüfungen. Wie die Literatursuche eine Schnittstelle darstellt und sich die Daten digitalisiert erheben lassen, zeigt dieser Beitrag.
Abkürzungen
CEP Clinical Evaluation Plan
CER Clinical Evaluation Report
CIP Clinical investigation plan
DiGA digitale Gesundheitsanwendung
MDR Medical Device Regulation; EU-Verordnung 2017/745
PMCF Post-market clinical follow-up
SotA State of the Art
Zugrundeliegende Regularien
EU-Verordnung 2017/745 (MDR)
Medizinprodukte-Durchführungsgesetzt (MPDG)
1. Einleitung
Klinische Daten sind für jeden Medizinproduktehersteller und für jedes Medizinprodukt unausweichlich:
„Klinische Daten“ bezeichnet Angaben zur Sicherheit oder Leistung, die im Rahmen der Anwendung eines Produkts gewonnen werden und die aus den folgenden Quellen stammen:
- klinische Prüfung(en) des betreffenden Produkts,
- klinische Prüfung(en) oder sonstige in der wissenschaftlichen Fachliteratur wiedergegebene Studien über ein Produkt, dessen Gleichartigkeit mit dem betreffenden Produkt nachgewiesen werden kann,
- in nach dem Peer-Review-Verfahren überprüfter wissenschaftlicher Fachliteratur veröffentlichte Berichte über sonstige klinische Erfahrungen entweder mit dem betreffenden Produkt oder einem Produkt, dessen Gleichartigkeit mit dem betreffenden Produkt nachgewiesen werden kann,
- klinisch relevante Angaben aus der Überwachung nach dem Inverkehrbringen, insbesondere aus der klinischen Nachbeobachtung nach dem Inverkehrbringen.
(QUELLE: MDR, Artikel 2)
Wie die klinischen Daten erhoben werden, sollte bereits zu Beginn jeder Produktentwicklung festgelegt werden. Im Auftrag der Hersteller erheben wir sie bei Bedarf über klinische Prüfungen oder über Literatursuchen, die wiederum auch als Basis für klinische Prüfungskonzepte, Evaluationskonzepte für DiGA-Studien und Beschreibungen des Stands der Technik (State oft he Art, SotA) dienen.
Zusammengefasst werden die erhobenen klinischen Daten in der klinischen Bewertung oder im Rahmen von deren Aktualisierung. Die klinische Bewertung ist somit im Zentrum einer jeden Datenerhebung oder anders ausgedrückt, jede Datenerhebung fließt in die klinische Bewertung ein, die diese Daten dann analysiert und beurteilt.
Dieser Beitrag zeigt unsere Lösung auf, klinische Daten für die klinische Bewertung oder als Input für eine klinische Prüfung digitalisiert und automatisiert zu erheben.
2. Typen klinischer Prüfungen
Bei Medizinprodukten wird zwischen vier verschiedenen klinischen Prüfungstypen unterschieden:
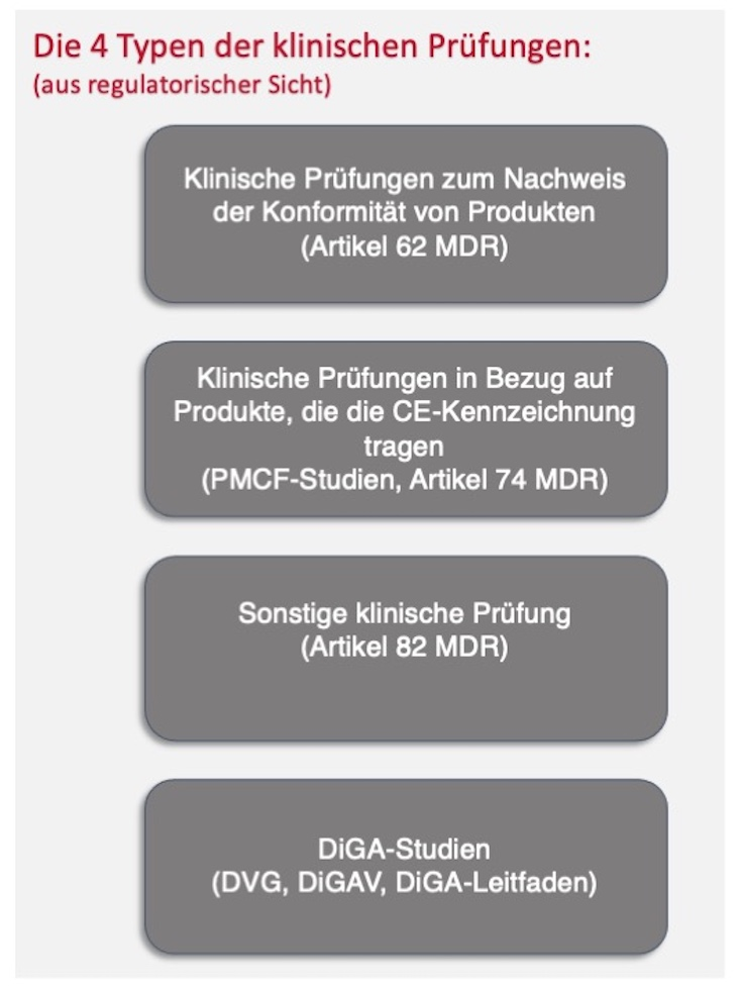
Abb. 1 Klinische Prüfungstypen
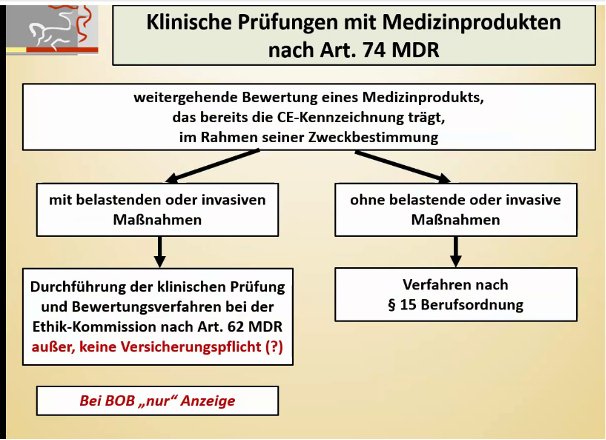
Im Fokus dieses Beitrags stehen insbesondere die klinische Prüfung zum Nachweis der Konformität von Produkten (Artikel 62 ff der MDR, auch „Zulassungsstudie“genannt), die klinische Prüfung in Bezug auf Produkte, die die CE-Kennzeichnung tragen (Artikel 74 der MDR oder Ausnahme davon, auch „PMCF-Studie“ genannt) sowie die DiGA-Studien.
Sonstige klinische Prüfungen (Artikel 82 der MDR) dienen vor allem dem wissenschaftlichen Erkenntnisgewinn und inden außerhalb des Konformitätsbewertungsverfahrens und außerhalb der klinischen Nachbeobachtung (Post Market Clinical Follow-up, PMCF) statt. Sie finden daher keinen Eingang in die klinische Bewertung. Nichtsdestotrotz muss aber auch für Prüfpläne dieser sonstigen Studien eine Literatursuche durchgeführt werden.
2.1 Klinische Prüfung zum Nachweis der Konformität von Produkten
Eine solche klinische Prüfung wird auch Zulassungsstudie genannt. In diesem Fall werden die klinischen Daten direkt zum Medizinprodukt im Rahmen einer klinischen Prüfung erhoben. Diese fließen dann neben State of the Art Daten in die initiale klinische Bewertung des Medizinproduktes mit ein.
Bei einer solchen klinischen Prüfung muss unter anderem eine präklinische Bewertung mit eingereicht werden. Diese enthält:
- präklinische/Verifizierungsdaten zum Produkt
- Daten zum Stand der Technik
Um an Daten zum Stand der Technik zu kommen, ist eine Literatursuche erforderlich.
Diese präklinische Bewertung dient gleichzeitig als Basis des klinischen Prüfplans (Clinical Investigation Plan, CIP), da dieser eine
„Beschreibung der Relevanz der klinischen Prüfung im Kontext des Stands der Technik bei der klinischen Praxis“
enthalten muss (Anhang XV, Kapitel II, Satz 3.4 der MDR).
Die im Rahmen dieser klinischen Prüfung im klinischen Prüfbericht (Anhang XV, Kapitel II, Satz 3.17 der MDR) zusammengefassten klinischen Daten fließen in die klinische Bewertung ein, die ebenfalls ein Kapitel zum Stand der Technik enthält, das aktualisiert aus der präklinischen Bewertung entnommen werden kann.
Die Literatursuche fokussiert sich hier auf klinische Daten zum Stand der Technik, alternative Verfahren und deren Outcomes.
2.2 PMCF-Studie
Bei klinischen Prüfungen in Bezug auf Produkte, die die CE-Kennzeichnung tragen (Artikel 74 der MDR) und davon ausgenommene PMCF-Studien muss ebenfalls ein Prüfplan erstellt werden, der – anders als bei Zulassungsstudien – auf der zuletzt erstellten klinischen Bewertung, deren möglichen Datenlücken und verbliebenen Fragestellungen basiert.
Auch hierzu wurde im Rahmen der klinischen Bewertung eine Literatursuche durchgeführt.
2.3 DiGA-Studien
DiGA-Studien dienen dem Nachweis des positiven Versorgungseffektes der digitalen Gesundheitsanwendung (DiGA).
Hierzu muss im Vorfeld des Fast-Track-Verfahrens ein nach allgemein anerkannten wissenschaftlichen Standards erstelltes Evaluationskonzept vorgelegt werden, das die Ergebnisse der systematischen Datenauswertung angemessen berücksichtigt.
„Die systematische Datenauswertung umfasst neben einer systematischen Literaturrecherche und -bewertung auch den Einschluss eigener systematisch ausgewerteter Daten, die in der Anwendung der DiGA gewonnen wurden.“
Somit muss auch im Kontext einer geplanten DiGA-Studie eine systematische Literatursuche durchgeführt werden, die dann auch in den CIP der DiGA-Studie einfließt.
Diese Literatur kann zum einen ansatzweise aus der klinischen Bewertung stammen. Da die Endpunkte der DiGA-Studie aber nicht nur die Claims der klinischen Bewertung zur klinischen Leistung, Sicherheit und zum klinischen Nutzen (über den Nachweis des medizinischen Nutzens der DiGA) abdecken können, sondern auch Endpunkte zu patientenrelevanten Struktur- und Verfahrensverbesserungen umfassen können, empfiehlt sich eine gesonderte Literatursuche.
3. Digitalisierte Literatursuche
Der vorherige Abschnitt zeigt:
Abb. 2 Literatursuche im Zentrum
Die Literatursuche kann mitunter sehr zeitintensiv und langwierig sein. Im Rahmen der Digitalisierung der Technischen Dokumentation wurden deshalb auch die klinische Bewertung und damit insbesondere die Literatursuche digitalisiert und der Prozess automatisiert.
Da es bei medXteam schwerpunktmäßig um klinische Daten geht, steht die Literatursuche im Mittelpunkt. Diesen Prozess haben wir über unseren Partner avasis als zertifizierter Smart Expert Partner der Siemens Digital Industries Software in den Bereichen Teamcenter und Polarion umgesetzt.
3.1 Digitalisierte Literatursuche über Polarion
Die Literatursuche ist der Kernprozess der klinischen Bewertung.
Bei der Literatursuche über Polarion wird eine direkte Verbindung zu den Datenbankquellen (z. B. direkt zu PubMed) hergestellt.
Die Literaturrecherche wird in Form der folgenden Dokumente durchgeführt und dokumentiert:
- Literatursuch- und Reviewplan (engl. Literature Search and Review Plan)
Der Literatursuch – und Review-Plan beschreibt die objektive Suche und beschreibt die Identifizierung von Publikationen. Er umfasst:
- Quellen der Publikationen
- Suchbegriffe
- definierte Filter
- Beurteilungskriterien und Prozess für identifizierte Publikationen
- Prozess zur Analyse der relevanten Publikationen
- Durchführungsprotokoll der Literaturrecherche (engl. Literature Search Execution Protocol)
Das Durchführungsprotokoll liefert Details zu den durchgeführten Recherchen und einen Überblick über die Historie der Recherchen. Es umfasst:
- verwendete Suchanfragen und Ergebnisse
- Abweichungen vom Literatursuch- und Reviewplan
- Übersicht über durchgeführte Recherchen und Suchergebnisse
- Bericht zur Literaturrecherche (engl. Literature Review Report)
Der Bericht enthält eine Zusammenfassung der durchgeführten Suche, sowie die Auswertung und Analyse. Er umfasst:
- Zusammenfassung der objektiven Suchdurchführung und -ergebnisse
- durchgeführte Suche und Auswahlverfahren zur Identifizierung mit anderen Mitteln
- Bewertung der identifizierten Publikationen
- Analyse der relevanten Publikationen (siehe nachfolgender Abschnitt)
Dokumentation der Analyse
Die Durchführung von objektiven Recherchen in Pubmed und Pubmed Central kann mit digitalen Softwarelösungen teilautomatisiert werden, um eine nachvollziehbare und reproduzierbare Recherchedokumentation zu gewährleisten sowie den Aufwand für die Dokumentation der Rechercheergebnisse zu reduzieren. Die eingesetzte Lösung (Polarion mit avaPubmed-Erweiterung) bietet eine direkte, validierte Schnittstelle zu Pubmed und Pubmed Central.
Der Volltext jeder potenziell relevanten Publikation wird gelesen und im Hinblick auf das Ziel (Scope) der Literaturrecherche und die relevanten klinischen Bewertungsthemen im jeweiligen klinischen Bewertungsplan analysiert. Die extrahierten Aussagen zu Sicherheit, Leistung, Nutzen, Anspruch oder Stand der Technik werden dokumentiert.
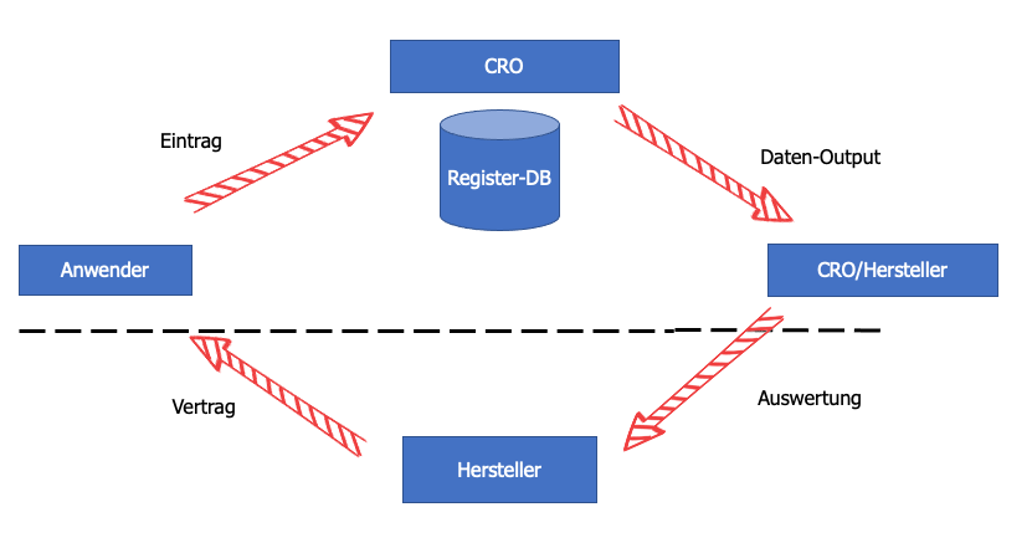
Die Analyse einer einzelnen "Publikation" wird in Form einer einzelnen "Publikationsbewertung" (engl. „Publication Evaluation“ siehe Schaubild unten) dokumentiert: Die "Publikation" ist mit der "Publikationsbewertung" verknüpft und die Bewertung ist mit dem jeweiligen "Klinischen Bewertungsgegenstand" im Klinischen Bewertungsplan verknüpft. Die folgende Grafik erläutert den Zusammenhang zwischen den einzelnen Workitem-Typen:
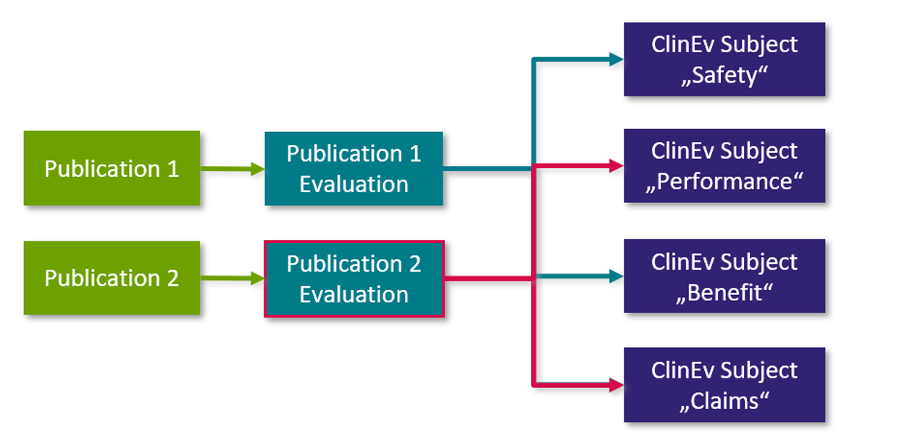
Abb. 3: Analyse
Bericht zur Literaturrecherche
Im Bericht zur Literaturrecherche wird eine Übersicht und Zusammenfassung der Analyse gegeben:
Es wird für jedes Klinische Bewertungsthema aufgeführt, welche Publikation mit Relevanz für dieses Thema identifiziert wurde und welche spezifischen Aussagen in der Publikationsbewertung extrahiert wurden.
Basierend auf diesen Ergebnissen wird analysiert, ob die relevanten Datensätze in ihrer Gesamtheit Evidenz für das jeweilige Clinical Evaluation Subject (den jeweiligen Claim, siehe Abbildung oben) zeigen. Das Ziel ist es, nach Konsistenz der Ergebnisse über bestimmte Klinische Bewertungsthemen hinweg zu suchen. Wenn unterschiedliche Ergebnisse über die Datensätze hinweg beobachtet werden, ist es hilfreich, den Grund für diese Unterschiede zu ermitteln.
Die folgende Grafik visualisiert den Zusammenhang der Dokumente und der enthaltenen digitalen Inhalte in Form von Workitems:
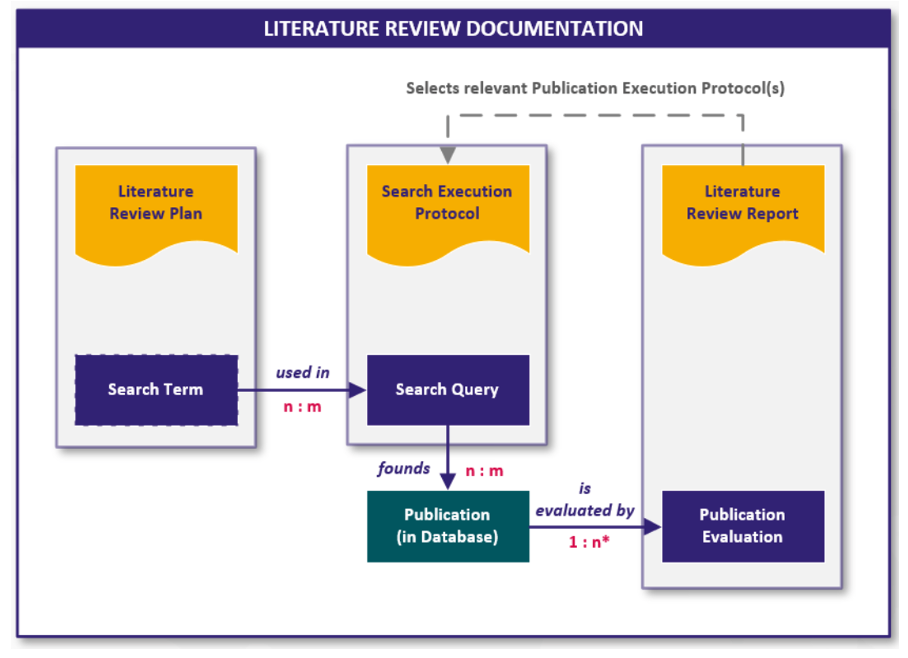
Abb. 4 Schnittstellen und Work Items
3.2 Digitalisierte klinische Bewertung
Die klinische Bewertung ist ein wesentlicher Bestandteil der Technischen Dokumentation (initial im Rahmen des Konformitätsbewertungsverfahrens und Aktualisierungen im Rahmen der klinischen Nachbeobachtung).
Ihr Kern ist die Literatursuche, die digitalisiert durchgeführt werden kann (s. o.). Eingebettet in Polarion als Subsystem kann auch sie selbst digitalisiert werden. Die folgende Abbildung gibt einen Überblick über den Inhalt der Dokumente zur klinischen Bewertung
- CEP,
- CER,
- Dokumente zur Literatursuche – Plan, Protokoll, Bericht
eingebettet als Subsystem in das Gesamtsystem Technische Dokumentation:
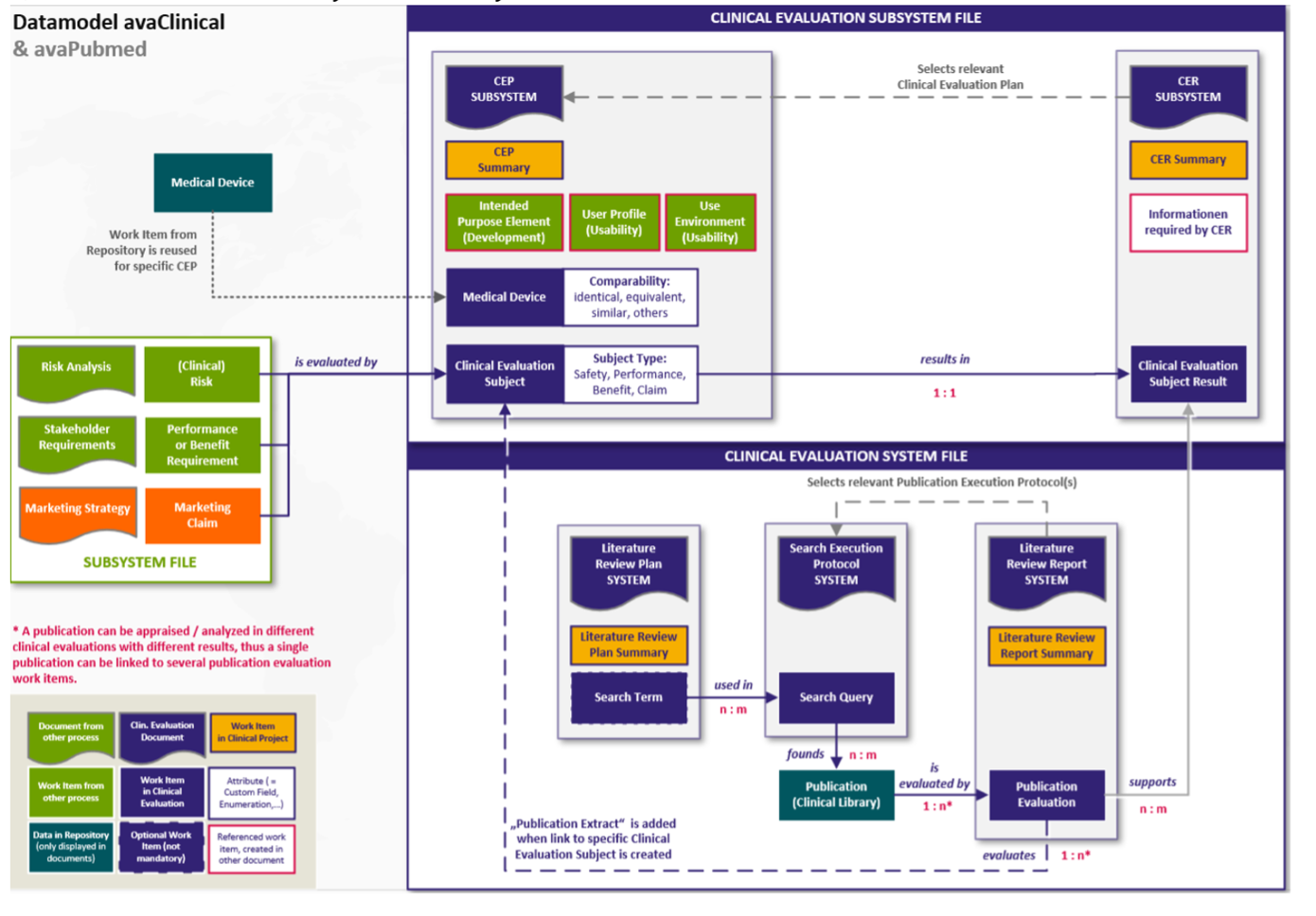
Abb. 5 Gesamtsystem Technische Dokumentation
3.3 Digitalisierte klinische Prüfung
Und mit der engen Verzahnung der klinischen Prüfung (egal ob zur Zulassung, im Rahmen der klinischen Bewertung oder im Rahmen einer DiGA-Studie) mit dem Prozess der Literatursuche und somit mit der klinischen Bewertung ist auch eine Digitalisierung der wesentlichen Dokumente der klinischen Prüfung wie z. B.
- Klinischer Prüfplan (Anhang XV, Kapitel II, Abschnitt 3 der MDR)
- Handbuch des klinischen Prüfers (Anhang XV, Kapitel II, Abschnitt 2 der MDR)
- präklinische Bewertung
möglich.
3.4 Vorteile der Digitalisierung
Die Digitalisierung der Technischen Dokumentation für Medizinprodukte und damit der klinischen Bewertung und der Literatursuche ist die Zukunft!
Die Vorteile der Digitalisierung liegen auf der Hand:
- effizienteres Arbeiten
- zielorientiertes Einsetzen der Kapazitäten
- Beseitigung von Ineffizienzen bei Erstellung, Pflege und Änderung von Inhalten der Technischen Dokumentation, klinischen Bewertung und Literatursuchen
- langfristige Verringerung des Pflegeaufwands
Über Polarion lassen sich Schnittstellen wie Zweckbestimmung, Risikomanagement, Gebrauchstauglichkeit, klinische Bewertung, klinische Prüfung Projekten zuordnen und bei Bedarf wiederverwenden. Die Erstellung und Pflege von Dokumenten wird somit deutlich vereinfacht und beschleunigt. Daneben werden Redundanzen und Inkonsistenzen vermieden.
4. Was wir für Sie tun können
Wenn eine klinische Prüfung durchgeführt werden soll, müssen zuvor grundlegende Sicherheits- und Leistungsanforderungen erfüllt und somit wesentliche Dokumente der Technischen Dokumentation erstellt werden.
Die klinische Prüfung mündet in der klinischen Bewertung, die dann wieder die Basis für PMCF-Aktivitäten (einschließlich einer PMCF-Studie) darstellt.
Wir unterstützen Sie deshalb während Ihrem kompletten Vorhaben mit Ihrem Medizinprodukt immer mit primärem Bezug auf die klinischen Daten zum Produkt: von Anfang an bis zum Ende.
Merken Sie sich hierzu bitte gerne zwei Veranstaltungen vor:
06.10.2021, 16:00 – 20:00 Uhr Start-up Night der Gesundheitswirtschaft
Fragestunde Regulatory & Clinical Affairs – Innovation und Technologie treffen auf Regulierung, Zulassung und Marktüberwachung (Hans Wenner, VDE und Daniela Penn, medXteam)
11.11.2021, 14:00 – 15:00 Uhr: Zweites kostenloses medXevent:
Digitalisierte klinische Bewertung im Blickwinkel klinischer Prüfungen
5. Wie wir Ihnen helfen können
Ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss, klären wir bei medXteam im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung.
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung