Das Thema „Real World Data“ und „Real World Evidence“ nimmt gerade durch digitale Gesundheitsanwendungen (DiGA) nun auch für Medizinprodukte Fahrt auf. Um was handelt es sich dabei? Und inwieweit lässt sich dieses Thema auf die Datenerhebung für Medizinprodukte übertragen? Wann macht es Sinn?
Zugrundeliegende Regularien
Digitale-Versorgung-Gesetz (DVG)
Digitale Gesundheitsanwendungen-Verordnung (DiGAV)
DiGA-Leitfaden
EU-Verordnung 2017/745 (MDR)
ISO 14155
1. Was sind Real World Data (RWD) und Real World Evidence (RWE)?
„Real World Data beziehen sich auf Daten über die Verwendung oder die potenziellen Vorteile oder Risiken eines Arzneimittels, das aus anderen Quellen als aus traditionellen klinischen Studien stammt.“ Diese Definition stammt von Jacqueline Corrigan-Curay, J.D., M.D., Direktorin des Office of Medical Policy-Centers der FDA. Er zeigt, dass dieses Thema bereits Einzug in die Arzneimittelbranche gehalten hat und insbesondere in den USA bereits Anwendung findet.
Was sind nun „Real World Data“? Damit bezeichnet man Datenerhebungen, die sich auf den tatsächlichen klinischen Routinealltag beziehen. Der Nachweis, der über diese Daten aus dem klinischen Routinealltag erbracht wird, wird als „Real World Evidence“ bezeichnet.
2. Real World Data – Erhebung und Nutzung
2.1 Real World Data bei Arzneimitteln
Real World Data werden in der Regel im Rahmen von Beobachtungsstudien erhoben. Diese sind für Arzneimittel reguliert. BfArM hat hierzu beispielsweise im Dezember 2019 folgende
„Gemeinsame Empfehlungen des Bundesinstituts für Arzneimittel und Medizinprodukte und des Paul-EhrlichInstituts zu Anwendungsbeobachtungen nach § 67 Absatz 6 Arzneimittelgesetz und zur Anzeige von nichtinterventionellen Unbedenklichkeitsprüfungen nach § 63f Arzneimittelgesetz“
veröffentlicht.
Solche Regulierungen gibt es bisher für Medizinprodukte nicht.
2.2 Daten aus dem klinischen Routinealltag bei Medizinprodukten
Bei DiGAs wird vor der DiGA-Studie oder dem Antrag auf Aufnahme in das DiGA-Verzeichnis ein Evaluationskonzept gefordert. Dieses soll eine „systematische Datenauswertung neben einer systematischen Literaturrecherche und -bewertung auch den Einschluss eigener systematisch ausgewerteter Daten, die in der Anwendung der DiGA gewonnen wurden,“ umfassen.
Somit sind dies Daten aus dem klinischen Routinealltag der Anwendung der DiGA.
Auch Roche Diabetes nimmt zu diesem Thema Stellung:
„Evaluierung des Nutzens digitaler Gesundheitsanwendungen über Real-World-Daten: Bei der Evaluierung des Nutzens digitaler Gesundheitsanwendungen sollte berücksichtigt werden, dass sich im Bereich der pharmakologischen Zulassungsverfahren zunehmend die Perspektive durchsetzt, dass randomisierte, kontrollierte Studien ein unvollständiges Abbild der Versorgungsrealität darstellen. Randomisierte, kontrollierte Studien sind dazu geeignet, valide Kausalitäten zwischen einer Intervention und ihrem Effekt herzustellen. Real-World-Daten (RWD) werden als potenzielle Quellen gesehen, um Einblicke darüber zu erhalten, wie zertifizierte Medizinprodukte und zugelassene Medikamente die Outcomes von Patienten in der realen Versorgung beeinflussen. Die europäische Arzneimittelbehörde (EMA) diskutiert deshalb intensiv, wie RWD zukünftig bei der Lösung komplexer Fragestellungen integriert werden können...“
(Quelle: Roche Diabetes Politikportal, Zugriff am 30.03.2021)
Die fortschreitende Digitalisierung des Gesundheitswesens und eine daraus resultierende ansteigende Verfügbarkeit von digitalen Datensätzen bilden die Grundlage für einen zukünftig intensiveren Einsatz von RWD und RWE. Diese Entwicklungen eröffnen potentielle Chancen für neue Player im System: Plattformen für einen Datenaustausch zwischen Leistungserbringern und Institutionen werden notwendig, um RWE Daten zu generieren und zu verarbeiten (Meinert et al., 2018).
Aber nicht nur das DVG fordert solche Daten, auch die MDR mit der klinischen Nachbeobachtung (Post-Market Clinical Follow-up, PMCF). Diese soll nämlich kontinuierlich klinische Daten zum Medizinprodukt erheben, und zwar mit dem primären Ziel zu prüfen, ob die Anwendung in der Normal- oder Routineversorgung für eine bestimmte Patienten oder Anwender wirksam ist. Diesen Daten müssen deshalb den Routinealltag und die Routineversorgung gut widerspiegeln.
In Anhang IXV der MDR heißt es in Satz 1 von Teil B:
„Die klinische Nachbeobachtung nach dem Inverkehrbringen ist als ein fortlaufender Prozess zur Aktualisierung der klinischen Bewertung gemäß Artikel 61 und Teil A dieses Anhangs zu verstehen und wird im Plan des Herstellers zur Überwachung nach dem Inverkehrbringen behandelt. Bei der klinischen Nachbeobachtung nach dem Inverkehrbringen sammelt und bewertet der Hersteller auf proaktive Weise klinische Daten, die aus der Verwendung eines die CE-Kennzeichnung tragenden, im Rahmen seiner Zweckbestimmung gemäß dem einschlägigen Konformitätsbewertungsverfahren in den Verkehr gebrachten oder in Betrieb genommenen Produkts im oder am menschlichen Körper hervorgehen, um die Sicherheit und die Leistung während der erwarteten Lebensdauer des Produkts zu bestätigen, die fortwährende Vertretbarkeit der ermittelten Risiken zu gewährleisten und auf der Grundlage sachdienlicher Belege neu entstehende Risiken zu erkennen.“
Da im klinischen Routinealltag die Bedingungen meist andere als bei einer randomisierten, kontrollierten klinischen Prüfung sind, die in einem festgelegten Rahmen stattfindet, eignen sich randomisierte, kontrollierte klinische Prüfungen (randomized controlled trial, RCT) nur bedingt als PMCF-Studie. Deren Ergebnisse lassen nur limitiert auf die eigentliche Routineanwendung übertragen. Außerdem können so auch nicht unbedingt neue Risiken und Chancen sowie ein Off-Label Use ermittelt werden.
2.3 Regulierung bei Medizinprodukten?
Wie lassen sich solche Studien aber nun regulatorisch in Bezug auf Medizinprodukte einordnen? Hier sollte zunächst ein Exkurs in die evidenzbasierte Medizin gemacht werden.
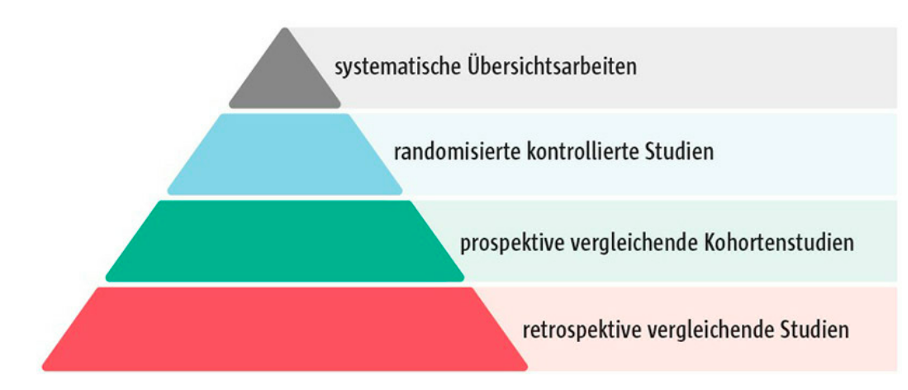
Abbildung 1: Evidenzhierarchie nach evidenzbasierter Medizin (EbM), Quelle: DiGA Vademecum)
Zunächst wird dabei zwischen interventionellen und nicht-interventionellen Studien, sogenannten Beobachtungsstudien, unterschieden. Wird bei interventionellen Studien die Anwendung des Medizinproduktes bei einer bestimmten Population geplant und durchgeführt und sind alle Bedingungen dazu festgelegt, spricht man von einer interventionellen Studie. Ergebnisse sind hier immer auf die Intervention zurückzuführen. Interventionelle Studien sind somit oft vergleichend und stets prospektiv. Zu den Interventionsstudien gehört die vielzitierte, vielgeforderte und wohl vielgefürchtete randomisierte kontrollierte Studie (Randomized Controlled Trial - RCT), der „Goldstandard" in der evidenzbasierten Medizin.
In Beobachtungsstudien wird keine geplante Intervention durchgeführt, sie werden daher auch nicht-interventionelle Studien genannt. Hier wird die Anwendung und der weitere Verlauf beim Patienten beobachtet und es werden entsprechende Schlussfolgerungen gezogen.
Bei Beobachtungsstudien wird also grundsätzlich keine Intervention gemäß klinischem Prüfplan durchgeführt, die Behandlung erfolgt ausschließlich nach der therapeutischen Praxis. Auch Beobachtungsstudien können sowohl vergleichend als auch nicht vergleichend durchgeführt werden; zudem können sie auch auf retrospektiven Daten basieren. Zu den bekanntesten nicht interventionellen Typen mit Kontrollgruppe gehören die Kohortenstudie und die Fall-Kontroll-Studie. Aber auch Register erheben Daten aus dem klinischen Routinealltag und werden anschließend retrospektiv ausgewertet.
Da die Ergebnisse von Beobachtungsstudien durch eine ganze Reihe von Verzerrungen (Bias) und Störfaktoren (Confounder) beeinflusst werden können, ist ihre interne Validität geringer als diejenige von Interventionsstudien. Ihre Evidenz ist jedenfalls in Bezug auf die Beantwortung der Frage nach dem klinischen Effekt einer konkreten Intervention grundsätzlich geringer als bei einer Interventionsstudie, da diese gerade die interne Validität bewertet. (Amboss, 2020)
Durch Beobachtung lassen sich Korrelationen feststellen; ein kausaler Zusammenhang ist damit jedoch nicht nachweisbar. Beobachtungsstudien sind im Vergleich zu Interventionsstudien in der Regel allerdings schneller und kostengünstiger durchführbar und verfügen im Vergleich zu Interventionsstudien über eine höhere externe Validität. Ohne den festgelegten Rahmen für die zu evaluierende Anwendung hat die Beobachtungsstudie zwar eine geringere interne Validität (und damit geringere Aussagekraft bzgl. der Wirksamkeit (Efficacy)), kann somit aber aber einen besseren Einblick in die Wirksamkeit im Rahmen der tatsächlichen Gegebenheiten des klinischen Routinealltags geben.
Bei diesen so erhobenen Daten handelt es sich um „Echtweltdaten" (Real World Data – RWD). Die daraus gewonnene Evidenz wird entsprechend als „Real World Evidence" (RWE) genannt.
Regulatorisch gesehen lässt sich das Medizinprodukt nur im klinischen Routinealltag anwenden, wenn es mit einem CE-Zeichen versehen ist. Der Beobachtungsstudie liegt kein klinischer Prüfplan zugrunde, sondern ein Beobachtungsplan. Somit trifft Artikel 74 der MDR nicht zu (§ 74 ist die Basis für klinische Prüfungen nach dem Inverkehrbringen, für die dennoch die in Anhang XV Kapitel II geforderten Dokumente erstellt werden müssen, z. B. der Prüfplan).
Bisher waren Beobachtungsstudien über § 23b MPG (Ausnahmen zur klinischen Prüfung) und der berufsrechtlichen Beratung nach § 15 der Berufsordnung für Ärzte (BO) reguliert. Dieser Paragraph fällt mit der MDR nun weg. Die MDR verweist in §82 (2) auf die Option der Mitgliedsstaaten, sonstige klinische Prüfungen auf lokaler Ebene zu regeln. Das deutsche Medizinprodukte-EU-Anpassungsgesetz – MPEUAnpG tut das, in dem es „sonstige klinische Prüfungen, die bereits das CE-Zeichen tragen in § 47 definiert. Dort ist auch klar formuliert, dass weder eine Anzeige bei der Bundesbehörde noch ein zustimmendes Votum der Ethikkommission von Nöten ist, wenn die Beobachtungsstudie die beiden folgenden Kriterien erfüllt:
- die Teilnehmer werden keinen zusätzlichen (zur therapeutischen Routinebehandlung) Belastungen/Therapien ausgesetzt
- das Medizinprodukt wird im Rahmen seiner Zweckbestimmung verwendet.
Was bleibt ist somit eine berufsrechtliche Beratung nach § 15 BO des Arztes, der die Beobachtungsstudie mit dem CE-gekennzeichneten Produkt gemäß Beobachtungsplan durchführt.
3. Was wir für Sie tun können
Da eine solche Datenerhebung von RWD ab dem 26. Mai 2021 nicht mehr reguliert ist und nicht unter das Dach der MDR fällt, bietet sie eine weitere Möglichkeit der Datenerhebung, um wiederum den P(ost) M(arket) C(linical) F(ollow-up)-Anforderungen der MDR gerecht zu werden.
Wir unterstützen Hersteller nicht nur beim Finden der richtigen Erhebungsmethode, sondern können auch in allen Punkten der Durchführung einer RWD-Beobachtungsstudie zur Seite stehen.
4. Wie wir Ihnen helfen können
Ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss, klären wir bei medXteam im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung.
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung
Literaturquellen
Amboss (2020) Studientypen der medizinischen Forschung. URL: https://www.amboss.com/de/wissen/ Studientypen der medizinischen Forschung (Zugriff am 30.03.2021)
Meinert E, Alturkistani A, Brindley D, Knight P, Wells G, Pennington N. The technological imperative for value-based health care. British Journal of Hospital Medicine. 2018;79(6):328-32