Die Blogreihe der medXteam GmbH geht im neuen Jahr weiter und greift mit dem ersten Beitrag 2021 das Thema DiGA-Studien auf.
Zugrundeliegende Regularien
Digitale-Versorgung-Gesetz (DVG)
Digitale Gesundheitsanwendungen-Verordnung (DiGAV)
DiGA-Leitfaden
1. Was ist eine DiGA?
Der Leitfaden gibt in Kapitel 2.1 eine Definition der „digitalen Helfer in den Händen der Patienten“. Demnach sind digitale Gesundheitsanwendungen (DiGAs) „Medizinprodukte der Risikoklasse I oder IIa (nach MDR oder, im Rahmen der Übergangsvorschriften bzw. bis zum Geltungsbeginn der MDR am 26.05.2021, nach MDD). Dabei beruht
- die Hauptfunktion der DiGA auf digitalen Technologien.
- Die DiGA ist keine digitale Anwendung, die lediglich dem Auslesen oder Steuern eines Gerätes dient; der medizinische Zweck muss wesentlich durch die digitale Hauptfunktion erreicht werden.
- Die DiGA unterstützt die Erkennung, Überwachung, Behandlung oder Linderung von Krankheiten oder die Erkennung, Behandlung, Linderung oder Kompensierung von Verletzungen oder Behinderungen.
- Die DiGA dient nicht der Primärprävention (siehe auch Kapitel 2.1.4 DiGA in der Prävention).
- Die DiGA wird vom Patienten oder von Leistungserbringer und Patient gemeinsam genutzt, d. h. Anwendungen, die lediglich vom Arzt zur Behandlung der Patienten eingesetzt werden („Praxisausstattung“), sind keine DiGA.“
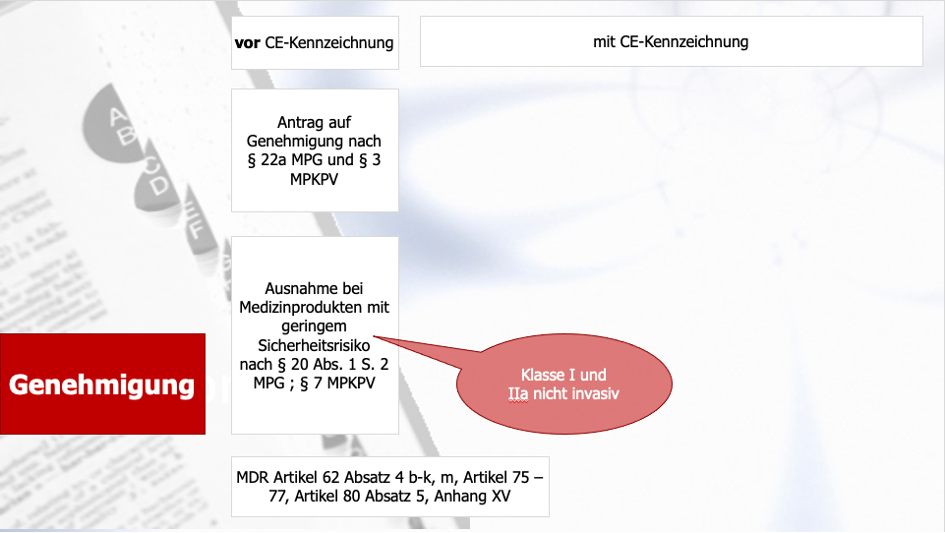
DiGA sind somit zugelassene Medizinprodukte, die ein CE-Zeichen tragen und somit die grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I der MDR erfüllt haben. Allerdings nur die Medizinprodukte der Klasse I und Klasse IIa. Auch die, die durch die MDR von Klasse I auf Klasse IIa hochgestuft werden. Doch alle Medizinprodukte der Klasse IIb und III und die, die unter der Richtlinie 93/42/EWG (MDD) unter die Klasse IIa fallen und mit der MDR in Klasse IIb und höher eingestuft werden, gehören nicht zu der Gruppe der DiGAs. Für diese kann keine Aufnahme in das Verzeichnis erfolgen.
2 Wie kommt die DiGA in das Erstattungsverzeichnis?
Das DiGA-Verfahren ist grundsätzlich nur mit einem CE-gekennzeichneten Produkt möglich. Der Hersteller kann nun entscheiden, ob er direkt und endgültig in das Verzeichnis aufgenommen werden möchte oder ob dies zunächst vorläufig geschehen soll.
Das Verfahren ist als sogenanntes „Fast-Track-Verfahren“ konzipiert.
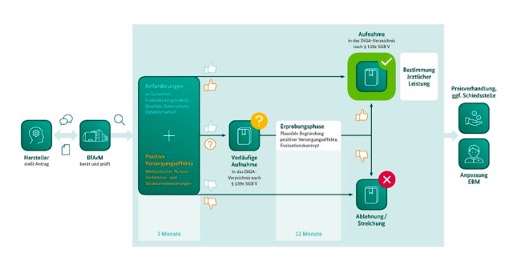
Bild1-DiGA: Ablauf des Fast-Track-Verfahrens. Quelle: DiGA-Leitfaden von BfArM
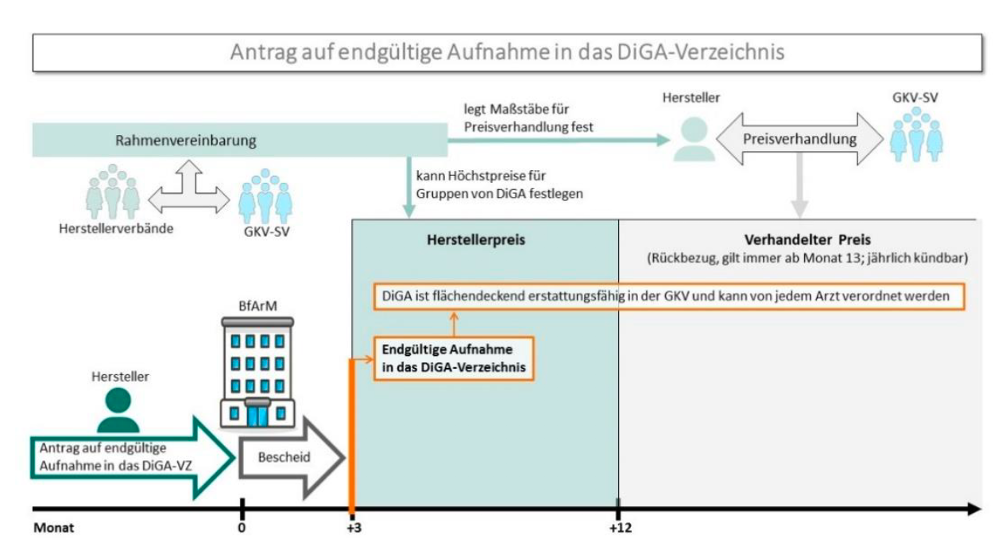
Bild2-DiGA: Antrag auf endgültige Aufnahme in das DiGA-Verzeichnis. Quelle: DiGA-Leitfaden von BfArM
Um als DiGA in das Erstattungsverzeichnis (DiGA-Verzeichnis) aufgenommen zu werden, sind verschiedene Anforderungen zu erfüllen und das Prüfverfahren beim BfArM muss erfolgreich durchlaufen werden. Dazu gehören unter anderem ein Evaluationskonzept und eine darauf aufbauende klinische Studie. Was bedeutet dies für die besagten Medizinprodukte? Wie können die Anforderungen erfüllt werden und wie kann das Verfahren am besten abgewickelt werden?
2.1 Was ist eine DiGA-Studie?
Neben den allgemeinen Anforderungen
- Sicherheit und Funktionstauglichkeit
- Datenschutz
- Informationssicherheit
- Interoperabilität
und weiteren Qualitätsanforderungen wie:
- Robustheit
- Verbraucherschutz
- Nutzerfreundlichkeit
- Unterstützung der Leistungserbringer
- Qualität der medizinischen Inhalte
- Patientensicherheit
muss der Hersteller einer DiGA nachweisen, welche positiven Versorgungseffekt realisiert werden. Der DiGA-Leitfaden definiert den positiven Versorgungseffekt folgendermaßen:
„Besonderer Schwerpunkt liegt, wie in der Definition der DiGA gemäß § 33a SGB V bereits angelegt, auf der Patientenzentrierung der nachzuweisenden Effekte. Sowohl medizinischer Nutzen als auch patientenrelevante Struktur und Verfahrensverbesserungen beziehen sich unmittelbar auf die Patienten und sind mittels entsprechender Endpunkte nachzuweisen.“
Ein medizinischer Nutzen (mN) ist demnach:
- eine Verbesserung des Gesundheitszustands (z. B. Reduzierung von Schmerzen, Verbesserung von Symptomen, …),
- eine Verkürzung der Krankheitsdauer (z. B. verkürzte Dauer der Krankschreibung, verkürzte Therapiedauer, …),
- eine Verlängerung des Überlebens oder
- eine Verbesserung der Lebensqualität.
Patientenrelevante Struktur und Verfahrensverbesserungen (pSVV) sind:
- Koordination der Behandlungsabläufe,
- Ausrichtung der Behandlung an Leitlinien und anerkannten Standards,
- Adhärenz,
- Erleichterung des Zugangs zur Versorgung,
- Patientensicherheit,
- Gesundheitskompetenz,
- Patientensouveränität,
- Bewältigung krankheitsbedingter Schwierigkeiten im Alltag
oder
- Reduzierung der therapiebedingten Aufwände und Belastungen der Patienten und ihrer Angehörigen.
2.2 Anforderungen an eine DiGA-Studie
Der Gesetzgeber stellt an eine DiGA-Studie besondere und klar definierte Anforderungen. Diese werden im DiGA-Leitfaden beschrieben:
- Grundsätzlich ist eine klinische Studie durchzuführen, Publikationen alleine reichen nicht aus.
- Der Hersteller muss in dieser Studie mindestens einen positiven Versorgungseffekt, der entweder aus dem Bereich des medizinischen Nutzens oder aus dem Bereich der patientenrelevanten Struktur und Verfahrensverbesserungen kommt, nachweisen.
- Zunächst sind die Patientengruppe und somit die Indikationen für die DiGA, für welche die Aufnahme ins DiGA-Verzeichnis beantragt wird, festzulegen. Nur für diese Indikationen erfolgt dann die Erstattung. Gemäß Leitfaden müssen die Definition und Eingrenzung dieser Patientengruppe „anhand einer oder mehrerer Indikationen nach ICD-10 erfolgen, wobei ausschließlich sowohl drei- als auch vierstellige Angaben zulässig sind.“
- Die Studie muss eine Überlegenheitsstudie sein, da sie zeigen muss, dass die Anwendung der DiGA besser ist als die Nichtanwendung. Deshalb handelt es sich um eine kontrollierte klinische Studie: Die Auswahl der Vergleichs- oder Kontrollgruppe muss dabei an der Versorgungsrealität orientiert sein. Beim Vergleich mit einer Behandlung ohne Anwendung einer DiGA ist z. B. auch ein Vergleich mit der Standardbehandlung (dem Standard of Care) möglich. Oder der Vergleich versus Nichtbehandlung bietet sich dann an, wenn eine DiGA zum Beispiel eine Versorgung für Patientinnen und Patienten anbietet, die andernfalls in der Mehrzahl unbehandelt blieben und ggf. auf einen Therapieplatz warten würden.
- Bei der Studie muss es sich um eine quantitative vergleichende Studie handeln und die gewählte Methodik muss adäquat zum gewählten Untersuchungsgegenstand sein. Folgenden Designs sind möglich:
- beobachtende/analytische Studie: z. B. Fall-/Kontrollstudien, Kohortenstudien
- experimentelle Interventionsstudie: z. B. nichtrandomisierte/randomisierte kontrollierte Studien
- Metaanalysen in Auswertung auch eigener Primärdaten
- Die DiGA-Studie kann einen prospektiven oder retrospektiven Ansatz haben. Letzteres beispielsweise dann, wenn das Medizinprodukt schon lange auf dem Markt ist und die passenden Daten in der geforderten Form (vergleichend) bereits mit der DiGA erhoben und entsprechend dokumentiert wurden).
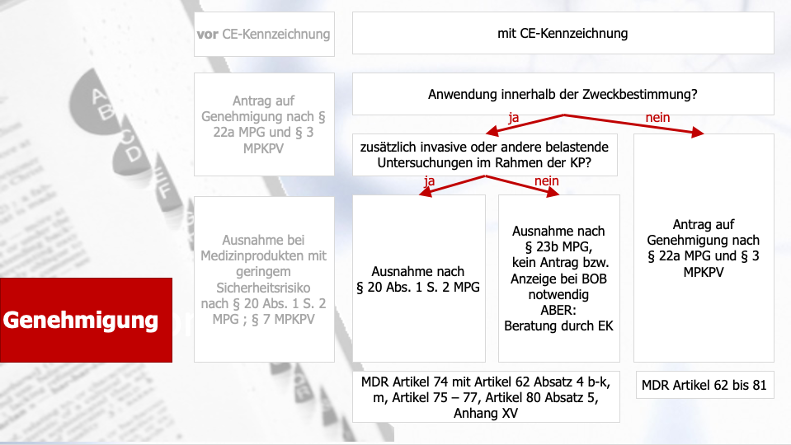
- Die DiGA-Studie muss in Deutschland durchgeführt werden: Entweder als PMCF-Studie, wenn das Medizinprodukt bereits zugelassen ist (Artikel 74 der MDR oder bis Mai 2021: § 23b MPG) ) oder als Zulassungsstudie zum Nachweis der Konformität des Medizinproduktes mit den grundlegenden Leistungs- und Sicherheitsanforderungen (Artikel 62 der MDR oder bis Mai 2021: §§ 20 – 23a MPG).
- Die DiGA-Studie muss weiterhin in ein Studienregister eingetragen werden und die Ergebnisse sind vollständig zu veröffentlichen
- Für die DiGA-Studie sind folgende Regularien für klinische Prüfungen mit Medizinprodukten anzuwenden:
- DIN EN ISO 14155 „Klinische Prüfung von Medizinprodukten an Menschen – Gute Klinische Praxis“ und die Richtlinie der FDA „Design Considerations for Pivotal Clinical Investigations for Medical Devices“
- Bei einer ärztlichen Beteiligung gelten die ethischen Grundsätze der Deklaration von Helsinki.
- Es muss mindestens eine berufsrechtliche Beratung bei einer Ethik-Kommission durchgeführt werden (siehe PMCF-Studie - § 23b MPG!) oder unter der MDR mindestens dann eine Stellungnahme der Ethikkommission eingeholt werden (Artikel 74 der MDR).
Das zeigt die Schnittstelle zu den Medizinprodukte-Regularien und die mögliche Nutzung dieser so erhobenen klinischen Daten für das PMCF (oder für die Zulassung des Medizinprodukts. Es empfiehlt sich deshalb dringend die Einhaltung der ISO 14155 und der MPG-/MDR-Anforderungen.
3. Was wir für Sie tun können
Eine DiGA-Studie ist eine nationale Besonderheit, schon alleine deshalb, weil sie nur in Deutschland durchführt werden kann. Es ist auch eine Studienanforderung an Medizinprodukte, für die normalerweise bzw. in der Regel im Rahmen der Erfüllung der grundlegenden Sicherheits- und Leistungsanforderungen für Medizinprodukte bei deren Nachweis in der klinischen Bewertung auf klinische Daten verzichtet werden kann. Stattdessen werden Leistungsdaten herangezogen.
Grundsätzlich holen wir DiGA-Hersteller dort ab, wo sie stehen und wir versuchen, regulatorische Medizinprodukte- mit DiGA-Anforderungen im Hinblick auf die klinische Studie möglichst miteinander zu verbinden, da ein solcher Aufwand durchaus für beide Bereiche genutzt werden kann. Somit können zwei Fliegen(MDR und DVG) mit einer Klappe geschlagen werden. Das fängt z. B. bei der Formulierung der richtigen Zweckbestimmung des Medizinprodukts an, um später bei Verhandlungen mit der Krankenkasse auch punkten zu können. Es geht mit der Einschätzung des richtigen Zeitpunkts der DiGA-Studie, mit dem Evaluationskonzept und der Studienplanung weiter und endet mit dem Nachweis des positiven Versorgungseffektes.
Deshalb erarbeiten wir mit den DiGA-Herstellern zunächst eine Strategie, wie sie auf ihrem Versorgungspfad nun optimal den positiven Versorgungseffekt nachweisen können. Und zwar abhängig von ihrer Ausgangssituation und von ihren Zielen.
4. Ausblick
Im nächsten Blogbeitrag werden wir uns einem wesentlichen Bestandteil der Planungsphase einer klinischen Prüfung, der statistischen Fallzahlplanung, detailliert zuwenden.
5. Wie wir Ihnen helfen können
Ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss, klären wir bei medXteam im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung.
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung