Im Rahmen unseres Weihnachtsspezials hatten wir noch im Teil 2 über das Antragsverfahren und den Genehmigungsprozess für klinische Prüfungen mit CE-gekennzeichneten Produkten geschrieben und sind davon ausgegangen, dass dies sowie der Artikel 74 für sämtliche PMCF-Studien mit bereits in Verkehr gebrachten Medizinprodukten gilt. Die virtuelle Veranstaltung von BfArM zusammen mit dem Bundesministerium für Gesundheit und dem Arbeitskreis der Ethik-Kommissionen brachte im Rahmen einer virtuellen Veranstaltung nun Erleuchtung zu diesem Artikel, was die Regelung zumindest in Deutschland angeht.
Abkürzungen
BOB (Bundesoberbehörde)
EK (Ethikkommission)
KP (klinische Prüfung)
MDR (medical device regulation; EU-Verordnung 2017/745)
MPG (Medizinproduktegesetzt)
MPAnpG-EU (Medizinprodukteanpassungsgesetz)
MPDG (Medizinproduktedurchführungsgesetz)
BO (Berufsordnung der Ärzte)
Zugrundeliegende Regularien
EU-Verordnung 2017/745 (MDR)
MPEUAnpG (das Medizinprodukte-EU-Anpassungsgesetz wurde am 25.05.2020 vom Bundestag als Gesetz verabschiedet. Dieses MPAnpG-EU beschreibt im Artikel 1 das Medizinprodukte-Durchführungsgesetz (MPDG))
MPDG (das MPDG wird das Medizinproduktegesetz (MPG) ab 26. Mai 2021 schrittweise ablösen und für alle Hersteller und Betreiber von Medizinprodukten in Deutschland rechtsverbindlich sein).
1. Einleitung
Seit 2017 ist die europäischen Medical Device Regulation (MDR) nun schon in Kraft und ihr Geltungsbeginn war bereits für den 26.05.2020 vorgesehen. Coronabedingt wurde dieser Geltungsbeginn sowie der des Medizinprodukterecht-Durchführungsgesetzes (MPDG) auf den 26. Mai 2021 verschoben. Damit ändern sich bekanntermaßen auch viele rechtliche Vorgaben und praktische Rahmenbedingungen für die Genehmigung und Durchführung klinischer Prüfungen von Medizinprodukten. Darüber informierte das BfArM am 05. Mai 2021 gemeinsam mit dem Bundesministerium für Gesundheit und dem Arbeitskreis der Ethik-Kommissionen im Rahmen einer virtuellen Veranstaltung.
Davon ausgehend, dass der Artikel 74 der MDR:
„Klinische Prüfungen in Bezug auf Produkte, die die CE-Kennzeichnung tragen
- Wird eine klinische Prüfung durchgeführt, die der weitergehenden Bewertung eines Produkts, das bereits die CE-Kennzeichnung gemäß Artikel 20 Absatz 1 trägt, im Rahmen seiner Zweckbestimmung dient (im Folgenden „klinische Prüfung nach dem Inverkehrbringen“), und würden im Rahmen dieser Prüfung Prüfungsteilnehmer zusätzlichen Verfahren zu den bei normalen Verwendungsbedingungen des Produkts durchgeführten Verfahren unterzogen, und sind diese zusätzlichen Verfahren invasiv oder belastend, so unterrichtet der Sponsor die betreffenden Mitgliedstaaten mindestens 30 Tage vor Beginn der Prüfung über das in Artikel 73 genannte elektronische System. Der Sponsor übermittelt die Unterlagen gemäß Anhang XV Kapitel II als Teil der Mitteilung. Für klinische Prüfungen nach dem Inverkehrbringen C1 gelten Artikel 62 Absatz 4 Buchstaben b bis k und m, Artikel 75, 76 und 77 und Artikel 80 Absätze 5 und 6 sowie die einschlägigen Bestimmungen des Anhangs XV.
- Wird eine klinische Prüfung durchgeführt, die der Bewertung eines Produkts, das bereits die CE-Kennzeichnung gemäß Artikel 20 Absatz 1 trägt, außerhalb seiner Zweckbestimmung dient, so gelten die Artikel 62 bis 81.“
für alle Medizinprodukte gilt, die das CE-Zeichen tragen, ging die Mehrheit davon aus, dass ab dem 26.05.2021 ein Ethikvotum für PMCF-Studien erforderlich ist und es keine berufsrechtliche Beratung nach § 15 der Berufsordnung für Ärzte (BO) mehr gibt. Dies ist nun nicht der Fall.
2. Verschiedene Arten der PMCF-Studien
2.1 Definition PMCF-Studien
Eine eigentliche Definition der PMCF-Studien gibt es nicht. Weder bisher in Richtlinie, MPG oder in einer der Verordnungen oder MEDDEVs, noch in der MDR oder im MPDG. Die MDR spricht in Artikel 74 nur davon, dass eine solche klinische Prüfung „klinische Prüfung nach dem Inverkehrbringen“ genannt wird:
„Wird eine klinische Prüfung durchgeführt, die der weitergehenden Bewertung eines Produkts, das bereits die CE-Kennzeichnung gemäß Artikel 20 Absatz 1 trägt, im Rahmen seiner Zweckbestimmung dient (im Folgenden „klinische Prüfung nach dem Inverkehrbringen“) […].“
Eine PMCF-Studie ist also eine klinische Prüfung, die mit dem CE-gekennzeichneten Produkt durchgeführt wird und klinische Daten zum Produkt im Rahmen der klinischen Nachbeobachtung (Post-Market Clinical Follow-up, PMCF) liefert. Die klinische Nachbeobachtung wird in der MDR in Anhang XIV Teil B geregelt.
Die klinische Nachbeobachtung umfasst aber nicht nur PMCF-Studien, sondern noch weitere mögliche Aktivitäten, um klinische Daten zum Produkt zu erheben. Ein Beispiel sind Real-World-Data, die im letzten Blog-Beitrag beschrieben wurden. Oder aber auch Registerdaten und weitere Aktivitäten. Die folgende Abbildung liefert einen Überblick über diese sowie über die gesamte Marktüberwachung (Post-Market Surveillance) gemäß MDR nach Artikel 83 - 85:

Abbildung 1. Allgemeine Methoden und Verfahren PMS und PMCF (Quelle: Keene A. Leveraging Post-Market Surveillance and Post-Market Clinical Follow-Up Data to Support EU Medical Device Regulation (MDR) Compliance, Whitepaper)
2.2 PMCF-Studien im Rahmen der Zweckbestimmung und ohne belastende Untersuchungen
Bisherige Regelung:
Bisher wurden diese PMCF-Studien gemäß § 23b MPG reguliert:
„§ 23b Ausnahmen zur klinischen Prüfung
Die §§ 20 bis 23a sind nicht anzuwenden, wenn eine klinische Prüfung mit Medizinprodukten durchgeführt wird, die nach den §§ 6 und 10 die CE-Kennzeichnung tragen dürfen, es sei denn, diese Prüfung hat eine andere Zweckbestimmung des Medizinproduktes zum Inhalt oder es werden zusätzlich invasive oder andere belastende Untersuchungen durchgeführt.“
Eine solche Studie fiel bisher unter die sonstigen Studien und lief außerhalb des MPG. Auf der Seite der Ethikkommission der Bayerischen Landesärztekammer sieht das z. B. so aus:
Abbildung 2: Studienarten der EK an der BLÄK
In Baden-Württemberg wird für eine solche Studie ein „freier Antrag“ gestellt:
Abbildung 3: Freier Antrag für eine solche Studie an der LÄK BW
Für PMCF-Studien innerhalb der Zweckbestimmung des Medizinprodukts war also eine berufsrechtliche Beratung nach § 15 der Berufsordnung für Ärzte erforderlich. Hierzu wurde bisher ein Antrag direkt an die Ethikkommission gerichtet. Manche Ethikkommissionen forderten hierzu x-fache Papierausfertigungen und eine Ausfertigung als CD-ROM. Andere wiederum (z. B. Hessen, Bayern) verfügen über ein Portal, über das die Anträge elektronisch hochgeladen werden können. Dann ist nur noch eine einfache Papierausfertigung erforderlich.
Für diese Studien muss neben bestimmten Studiendokumenten (Prüfplan, Patienteninformation und Einwilligungserklärung, Fragebögen, etc.) der Lebenslauf des Prüfers eingereicht werden. Eine Qualifikation, nachgewiesen über eine mindestens zweijährige Erfahrung des Prüfers mit klinischen Prüfungen mit Medizinprodukten, wie bei einem Ethikvotum erforderlich, wird hier nicht überprüft.
Regelung ab Geltungsbeginn der MDR
Bisher war unsere Annahme und Interpretation des Artikels 74 der MDR; dass hierunter alle PMCF-Studien fallen und somit dann ab 26.05.2021 ein Ethikvotum erforderlich sein würde.
Bei der o. g. Veranstaltung wurde nun der Artikel 74 für Deutschland folgendermaßen interpretiert:
- Er gilt nur für klinische Prüfungen mit CE-gekennzeichneten Produkten, die außerhalb ihrer Zweckbestimmung durchgeführt werden.
- Er gilt außerdem für klinische Prüfungen mit CE-gekennzeichneten Produkten, wenn in deren Rahmen zusätzliche belastende Untersuchungen durchgeführt werden.

Das bedeutet, dass es weiterhin das oben beschriebene Verfahren über sonstige Studien und freie Anträge (berufsrechtliche Beratung nach § 15 BO) gibt!
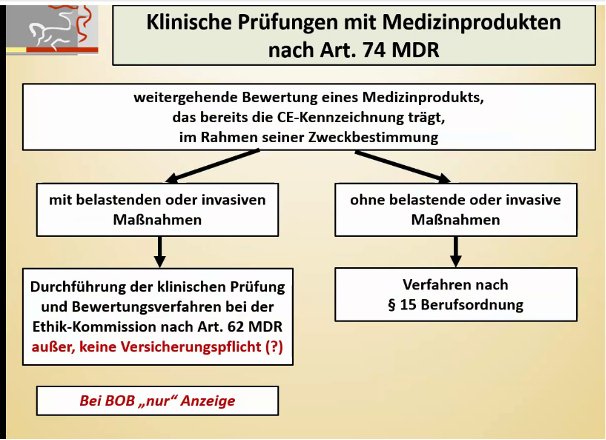
Abbildung 4: Artikel 74 der MDR (Quelle: Präsentationsfolien, BfArM-Veranstaltung, https://www.bfarm.de/DE/Service/Veranstaltungen/Dialogveranstaltungen/2021/210505-klinische_Pruefungen_von_MP.html)
Der Artikel 74 der MDR gliedert somit die PMCF-Studien innerhalb der Zweckbestimmung aus und betrachtet diese weiterhin wie schon im MPG als Ausnahme zu den klinischen Prüfungen.
Somit ändert sich hier bis auf Bezeichnungen im Prüfplan zu MPG und ggf. Richtlinie 92/43/EWG (Änderung in MDR, da nicht mehr gültig) nichts. Das Verfahren bleibt dasselbe und die Hersteller haben weiterhin die Möglichkeit, auf unkomplizierterem Weg ihre klinischen Daten im Rahmen der PMCF-Studie zu erheben.
2.3 PMCF-Studien mit belastenden Untersuchungen
Bisherige Regelung:
Bisher wurden diese PMCF-Studien ebenfalls gemäß § 23b MPG reguliert:
„§ 23b Ausnahmen zur klinischen Prüfung
Die §§ 20 bis 23a sind nicht anzuwenden, wenn eine klinische Prüfung mit Medizinprodukten durchgeführt wird, die nach den §§ 6 und 10 die CE-Kennzeichnung tragen dürfen, es sei denn, diese Prüfung hat eine andere Zweckbestimmung des Medizinproduktes zum Inhalt oder es werden zusätzlich invasive oder andere belastende Untersuchungen durchgeführt.“
Im Falle zusätzlicher belastender Untersuchungen fanden bisher dann bei einer solchen Studie zunächst wieder die § 20ff des MPG und in diesem Fall in Verbindung mit § 7 der MPKPV mit Abschnitt 1 Satz 3 Anwendung:
„Medizinprodukte, die nach den §§ 6 und 10 des Medizinproduktegesetzes die CE-Kennzeichnung tragen dürfen und deren klinische Prüfung zusätzliche invasive oder andere belastende Untersuchungen beinhaltet, es sei denn, diese Prüfung hat eine andere Zweckbestimmung des Medizinproduktes zum Inhalt.“
In diesem Fall war über das Medizinprodukteinformationssystem (MPI, ehemals DIMDI) ein Antrag bei BfArM auf Befreiung von der Genehmigungspflicht und bei der Ethikkommission ebenfalls über das MPI ein Antrag auf Stellungnahme (Votum) zu stellen.
Regelung ab Geltungsbeginn der MDR
Mit dem Geltungsbeginn der MDR wird dieses Verfahren durch das neue, im Artikel 74 der MDR geregelte, abgelöst:
- Der Sponsor unterrichtet die Bundesoberbehörde (BOB, in Deutschland: BfArM) mindestens 30 Tage vor Beginn der Prüfung über das MPI (in Deutschland).
- Es gelten Artikel 62 Absatz 4 Buchstaben b bis k und m, Artikel 75, 76 und 77 und Artikel 80 Absätze 5 und 6
- Es gelten außerdem die einschlägigen Bestimmungen des Anhangs XV
Das bedeutet, dass die BOB zu informieren und bei der Ethikkommission gemäß Artikel 62 Absatz 4 Buchstabe b ein Ethikvotum einzuholen ist.
Außerdem gelten im MPDG das Kapitel 4 mit den Abschnitten 1 und 2 und im letzteren mit Unterabschnitt 1 im Hinblick auf die Beantragung, Genehmigung und jeweiligen Fristen. Siehe hierzu auch der Blogbeitrag im Weihnachtsspezial Teil 2.
2.4 PMCF-Studien außerhalb der Zweckbestimmung
Bisherige Regelung:
Bisher wurden diese PMCF-Studien ebenfalls gemäß § 23b MPG reguliert:
„§ 23b Ausnahmen zur klinischen Prüfung
Die §§ 20 bis 23a sind nicht anzuwenden, wenn eine klinische Prüfung mit Medizinprodukten durchgeführt wird, die nach den §§ 6 und 10 die CE-Kennzeichnung tragen dürfen, es sei denn, diese Prüfung hat eine andere Zweckbestimmung des Medizinproduktes zum Inhalt oder es werden zusätzlich invasive oder andere belastende Untersuchungen durchgeführt.“
Bezog sich bisher die klinische Prüfung mit dem CE-gekennzeichneten Produkt auf eine neue Zweckbestimmung (z. B. somit auch auf neue Indikationen), so fanden bisher dann bei einer solchen Studie ebenfalls wieder die § 20ff des MPG Anwendung. Das heißt, es wurde eine klassische Zulassungsstudie gemäß den §§ 20ff des MPG durchgeführt:
- Antragsstellung über das MPI bei BfArM und EK
Regelung ab Geltungsbeginn der MDR
Mit dem Geltungsbeginn der MDR findet nun Artikel 74 Satz 2 Anwendung:
„(2) Wird eine klinische Prüfung durchgeführt, die der Bewertung eines Produkts, das bereits die CE-Kennzeichnung gemäß Artikel 20 Absatz 1 trägt, außerhalb seiner Zweckbestimmung dient, so gelten die Artikel 62 bis 81.“
Das bedeutet, es ist auch in diesem Fall eine klassische klinische Prüfung gemäß Artikel 62ff der MDR durchzuführen.
Außerdem gelten im MPDG das Kapitel 4 mit den Abschnitten 1 und 2 und im letzteren mit Unterabschnitt 1 im Hinblick auf die Beantragung, Genehmigung und jeweiligen Fristen. Siehe hierzu auch der Blogbeitrag im Weihnachtsspezial Teil 1:
Antrag auf Genehmigung einer klinischen Prüfung gemäß Artikel 70 Abs. 7 der MDR:
Verkürztes Verfahren Absatz 7a
Für Medizinprodukte ohne CE-Kennzeichnung und für CE-gekennzeichnete Produkte, wenn die klinische Prüfung außerhalb der Zweckbestimmung erfolgt (Klasse I und IIa nicht invasiv)
Der Antrag muss die Dokumente aus Anhang XV Kapitel II der MDR sowie die positive Stellungnahme der EK enthalten.
Volles Antragsverfahren Absatz 7b
Für Medizinprodukte ohne CE-Kennzeichnung und für CE-gekennzeichnete Produkte, wenn die klinische Prüfung außerhalb der Zweckbestimmung erfolgt (Klasse IIa invasiv, IIb* und III)
Der Antrag muss die Dokumente aus Anhang XV Kapitel II der MDR sowie die positive Stellungnahme der EK enthalten.
* Ausnahme in Deutschland, normalerweise gilt das verkürzte Verfahren auch für Produkte der Klasse IIb, nur in Deutschland gilt hier das volle Antragsverfahren.
3. Was wir für Sie tun können
Wir unterstützen zunächst dabei, für Sie die richtige Datenerhebungsmethode im Rahmen des PMCF zu finden. Soll eine PMCF-Studie durchgeführt werden, finden wir mit Ihnen den richtigen Weg der Umsetzung.
4. Wie wir Ihnen helfen können
Ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss, klären wir bei medXteam im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung.
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung