Im ersten Blog-Beitrag 2021 ging es bereits um das Thema „DiGA“ und die Datenerhebung. In diesem Beitrag möchten wir nun auf die Vorbereitungsaufgabe näher eingehen und deshalb geht es dieses Mal um das Evaluationskonzept. Dieses ist zusammen mit dem Prüfplan für die DiGA-Studie ebenfalls mit dem Antrag auf Aufnahme in das Erstattungsverzeichnis einzureichen. Was es damit auf sich hat, wie es am besten erstellt wird und was dabei alles zu beachten ist, erläutert dieser Blog-Beitrag.
Abkürzungen
BOB (Bundesoberbehörde)
BtB (Business-to-Business)
BtC (Business-to-Customer)
DiGA (digitale Gesundheitsanwendung)
MDR (medical device regulation; EU-Verordnung 2017/745)
Zugrundeliegende Regularien
Digitale-Versorgung-Gesetz (DVG)
Digitale Gesundheitsanwendungen-Verordnung (DiGAV)
DiGA-Leitfaden
EU-Verordnung 2017/745 (MDR)
ISO 14155
1. Einleitung
Um als DiGA in das Erstattungsverzeichnis (DiGA-Verzeichnis) aufgenommen zu werden, sind verschiedene Anforderungen zu erfüllen und das Prüfverfahren beim BfArM muss erfolgreich durchlaufen werden. Dazu gehören unter anderem, wenn noch keine Studie, die den DiGA-Kriterien entspricht, durchgeführt wurde, ein Evaluationskonzept und eine darauf aufbauende klinische Studie. Zur DiGA-Studie liefert der Januar-Blog-Beitrag wichtige Informationen.
Dieser Beitrag beschäftigt sich mit dem Evaluationskonzept für den positiven Versorgungseffekt der DiGA. Der DiGA-Leitfaden geht darauf in Kapitel 4.5.2 näher ein:
„Der Hersteller legt darüber hinaus mit dem Antrag ein nach allgemein anerkannten wissenschaftlichen Standards erstelltes Evaluationskonzept vor, das die Ergebnisse der systematischen Datenauswertung angemessen berücksichtigt. Das Studienprotokoll der angestrebten Studie soll ein Teil des Evaluationskonzepts sein. Die Wahl der Outcomes und des Studiendesigns des gewählten Vergleichs und der Versorgungsrealität sind zu begründen. Es ist darzustellen, warum und wie aus dem gewählten Evaluationskonzept die Nachweise der angestrebten pVE hervorgehen. Dieses muss von einem herstellerunabhängigen wissenschaftlichen Institut erstellt worden sein.“
„Das vorzulegende wissenschaftliche Evaluationskonzept soll gemäß § 15 DiGAV die Ergebnisse der systematischen Datenauswertung angemessen berücksichtigen.“
Auszug aus: Brönneke, Jan B. „DiGA VADEMECUM: Was man zu Digitalen Gesundheitsanwendungen wissen muss (German Edition).
Der Hersteller muss dieses somit nicht selbst erstellen, weil der Gesetzgeber die Erstellung im Leitfaden aber auch bereits im DVG durch ein wissenschaftliches unabhängiges Institut fordert. Dennoch trägt er einen wesentlichen Teil zur Erstellung bei, denn das Studienkonzept inklusive der zu belegenden Endpunkte für den positiven Versorgungseffekt bedürfen einer eingehenden Beschäftigung mit diesem Thema. Was das bedeutet, möchte nun dieser Beitrag näher erläutern. Gleichzeitig wird aufgezeigt, wie das Evaluationskonzept für eine sich bereits seit längerem auf dem Markt befindlichen DiGA (Software als Medizinprodukt) oder für eine sich z. B. gerade noch im Entwicklungsprozess befindlichen DiGA oder einer, die soeben noch unter der MDD zugelassen wurde, erstellt werden kann.
2. DiGA-Evaluationskonzept
In den DiGA-Regelwerken wird das Evaluationskonzept folgendermaßen definiert:
„Soll ein Antrag auf Erprobung gestellt werden, muss diesem ein wissenschaftliches Evaluationskonzept beigefügt werden. Dieses muss von einer herstellerunabhängigen Institution zum Nachweis des positiven Versorgungseffekts nach allgemein anerkannten wissenschaftlichen Standards erstellt werden.“
(Quelle: DiGA-Leitfaden)
Dieses umfasst insbesondere die folgenden Angaben zum geplanten Studienvorhaben, um den positiven Versorgungseffekt der DiGA nachzuweisen:
- die Angabe des Erprobungszeitraums (maximal 12 Monate)
- systematische Datenauswertung mit der DiGA selbst
- Beschreibung unter Nutzung des PICO-Schemas in der Kurzfassung des positiven Versorgungseffektes
- Konkretisierung der Patientengruppe durch die Angabe der entsprechenden ICD-Codes
- Art der positiven Versorgungseffekte der DiGA: medizinischer Nutzen und/oder patientenrelevante Verfahrens- und Strukturverbesserungen
- Angaben zu Forschungsdesign und Ergebnissen
- Angaben zur qualitätsgesicherten Anwendung der DiGA und zu Ausschlusskriterien
- Angaben zum einbezogenen wissenschaftlichen und herstellerunabhängigen Institut
Wir gliedern unser Evaluationskonzept beispielsweise folgendermaßen:

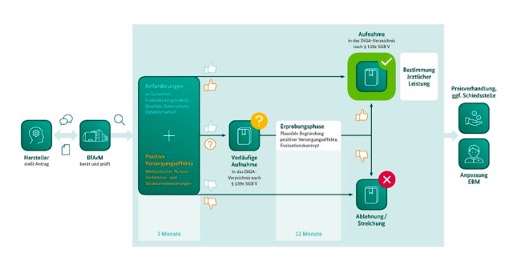
Abb. 1: Inhalt und Struktur des Evaluationskonzeptes
Ein wesentlicher Bestandteil des wissenschaftlichen Evaluationskonzepts ist eine systematische Datenauswertung im Rahmen der Anwendung der DiGA. Deshalb muss diese Datenauswertung mit dem zugelassenen Medizinprodukt durchgeführt werden. Das ist kein Problem, wenn die DiGA bereits auf dem Markt ist und Daten bereits durch ihre Anwendung erhoben wurden. Bei sich noch in der Entwicklung befindenden oder gerade zugelassenen Produkten können somit allerdings noch keine Daten für eine entsprechende Auswertung vorliegen. Eine solche Erhebungsphase ist deshalb im Anschluss an die Zulassung mit einzuplanen, bevor ein Antrag auf vorläufige Aufnahme in das Verzeichnis gestellt werden kann.
Unabhängig davon muss der Hersteller sich Gedanken über den mit der DiGA zu beschreitenden geplanten Versorgungspfad machen, für den der positive Versorgungseffekt nachgewiesen werden soll. Im Rahmen der Datenerhebung für das Evaluationskonzept sollten diverse Endpunkte bereits feststehen, die in diesem Zusammenhang auf Validität überprüft werden können.
Ziel und Zweck der Datenauswertung sollte sein, die Endpunkte der DiGA-Studie zu definieren, anhand derer der positive Versorgungseffekt auf dem vorgesehenen Versorgungspfad nachgewiesen werden kann. Sinn macht deshalb eine Auswahlmöglichkeit aus den Bereichen
- medizinischer Nutzen
- patientenrelevante Verfahrens- und Strukturverbesserungen
Wie sollen diese Daten aber nun erhoben werden, um dann für das Evaluationskonzept ausgewertet werden zu können?
2.1 Bereits zugelassene DiGA
Viele der bereits gelisteten DiGAs sind zugelassene Medizinprodukte, die bereits auf dem Markt waren. Somit besteht die Möglichkeit, eine bereits durchgeführte Studie heranzuziehen, die den DiGA-Kriterien entspricht, um sofort endgültig in das Verzeichnis aufgenommen zu werden. In diesem Fall empfiehlt sich unbedingt eine Beratung beim BfArM. Nämlich insbesondere dann, wenn Unsicherheit dahingehend besteht, ob die damit erhobenen Daten ausreichend sind und deshalb unklar ist, ob sie in die richtige Richtung gehen und ob damit ein Antrag auf eine vorläufige oder eine endgültige Aufnahme gestellt werden sollte.
Wurde noch keine Studie durchgeführt, bietet sich in diesem Fall an, Daten, die im Rahmen der Anwendung mit der DiGA erhoben wurden und über die App beim Hersteller vorliegen, retrospektiv auszuwerten.
Hinweis: Dies ist problemlos möglich, wenn zwischen Hersteller und Anwender eine BtC-Beziehung besteht. Gibt es diese nicht, weil die DiGA beispielsweise nicht direkt vom Hersteller, sondern z. B. von Therapeuten zur Verfügung gestellt wird (mit dem zum Hersteller eine BtB-Beziehung besteht), liegen die Daten nicht beim Hersteller.
Hat der Hersteller also Zugriff auf die Daten, die mit der DiGA im Rahmen der Anwendung automatisch erhoben werden, können diese anonymisiert ausgewertet werden. Dies geschieht in diesem Fall über einen sogenannten Beobachtungsplan, die Erhebung bezieht sich auf „Real World Data“ und da sie völlig anonym sind, können Sie ohne Einbeziehung einer Ethik-Kommission oder Behörde entsprechend gesammelt werden. Im Beobachtungsplan werden die zu erhebenden Parameter definiert, die sich auf die o. g. Aspekte
- medizinischer Nutzen
- patientenrelevante Verfahrens- und Strukturverbesserungen
beziehen sollten. Zu Real World Data empfiehlt sich auch unser Märzbeitrag „Medizinprodukte und Real World Data sowie Real World Evidence“ (Link: https://www.medxteam.de/index.php/medxteam-blog/15-medizinprodukte-und-real-world-data-sowie-real-world-evidence).
2.2 Noch nicht oder gerade erst zugelassene DiGAs
Mit diesen Produkten können noch keine eigenen Daten erhoben worden sein. In der Regel wird in diesem Fall auch die klinische Bewertung über Leistungsdaten (s. auch Artikel 61 Abschnitt 1 der MDR) erstellt, da auf klinische Daten bei diesen Klasse I oder IIa Produkten im Rahmen der Erfüllung der grundlegenden Sicherheits- und Leistungsanforderungen verzichtet werden kann.
Somit werden üblicherweise auch keine Zulassungsstudien durchgeführt. Leitlinien für State of the Art Kapitel in der klinischen Bewertung gibt es meist nur für die zugrundeliegenden Indikationen und alternative Anwendungsmöglichkeiten, da DiGAs eher innovativen Charakter haben und noch nicht umfassend in Leitlinien verankert sind.
Allerdings sind für die klinische Bewertung Claims zur klinischen Leistung, Sicherheit und zum klinischen Nutzen bereits im Plan für die klinische Bewertung zu definieren und dann im klinischen Bewertungsbericht mit Daten zu belegen.
Hinweis: Genau hier empfiehlt sich die Nutzung der Schnittstelle zum DiGA-Thema „medizinischer Nutzen“ bzw. patientenrelevante Struktur- und Verfahrensverbesserungen, denn diese Daten können dann nach der DiGA-Studie in der Aktualisierung der klinischen Bewertung genutzt werden.
Außerdem endet der DiGA-Prozess ja nicht mit der Listung im DiGA-Verzeichnis. Anschließend gehen die Verhandlungen mit den Krankenkassen los.
Hinweis: Deshalb am besten bei der Definition der Zweckbestimmung des Medizinprodukts sowie bei den Claims Schlagwörter aus dem medizinischen Nutzen oder zu patientenrelevanten Struktur- und Verfahrensverbesserungen, wenn möglich, einfließen lassen. Das erleichtert spätere Verhandlungen. Ebenfalls am besten nicht von „Software“ sondern von digitaler Gesundheitsanwendung sprechen.
Somit ist nach der Zulassung des Medizinprodukts die Datenerhebung für das Evaluationskonzept einzuplanen.
Auch hier empfiehlt sich eine Sammlung der mit der DiGA im Rahmen ihrer Anwendung beim Hersteller erhobenen Daten. Diese Erhebung ist nun aber nicht retrospektiv, sondern prospektiv in die Zukunft gerichtet.
Man kann aber auch nach einem definierten Anwendungszeitraum der DiGA die Daten retrospektiv auswerten.
Auch hier ist wichtig, dass die Daten
- mit der DiGA selbst und
- anonym
erhoben werden. Am besten funktioniert dies auch hier im BtC-Fall. Doch auch im BtB-Fall, wenn der Hersteller nicht direkt Zugriff auf die App-Daten hat, kann eine Beobachtungsstudie und Erhebung der Real World Daten erfolgen. Hier muss im Grunde nur die anonymisierte Bereitstellung im BtB-Verhältnis sichergestellt werden.
2.3 Zusammenfassung
Ein wesentlicher Aspekt des Evaluationskonzeptes sind Daten zum eingeschlagenen Versorgungspfad und dem dazu erforderlichen Nachweis des
- medizinischen Nutzens
- oder der patientenrelevanten Verfahrens- und Strukturverbesserungen.
Man definiert deshalb vorab Parameter, die nach der Datenerhebung entsprechend ausgewertet werden sollen. Diese sollten aus den folgenden Bereichen stammen:
Medizinischer Nutzen:
- Verbesserung des Gesundheitszustands,
- Verkürzung der Krankheitsdauer,
- Verlängerung des Überlebens oder
- Verbesserung der Lebensqualität
Patientenrelevante Struktur- und Verfahrensverbesserungen:
- im Rahmen der Erkennung, Überwachung, Behandlung oder Linderung von Krankheiten oder der Erkennung, Behandlung, Linderung oder Kompensierung von Verletzungen oder Behinderungen und
- auf eine Unterstützung des Gesundheitshandelns der Patientinnen und Patienten oder eine Integration der Abläufe zwischen Patientinnen und Patienten und Leistungserbringern ausgerichtet und
- umfassen insbesondere die Bereiche der
- Koordination der Behandlungsabläufe,
- Ausrichtung der Behandlung an Leitlinien und anerkannten Standards,
- Adhärenz,
- Erleichterung des Zugangs zur Versorgung,
- Patientensicherheit,
- Gesundheitskompetenz,
- Patientensouveränität,
- Bewältigung krankheitsbedingter Schwierigkeiten im Alltag oder
- Reduzierung der therapiebedingten Aufwände und Belastungen der Patienten und ihrer Angehörigen.
3. Was wir für Sie tun können
Wir fungieren als wissenschaftliche herstellerunabhängige Institution (CRO). Als solche erstellen wir Ihr Evaluationskonzept und beraten Sie auch schon in Ihrer frühen Entwicklungsphase schnittstellenkonform hinsichtlich Ihrer Claims zum Medizinprodukt oder Festlegung der Zweckbestimmung. All das geschieht mit den DiGA-Anforderungen im Visier, sodass Sie mit allem zwei Fliegen mit einer Klappe schlagen können.
Steht Ihre technische Dokumentation bereits, schauen wir uns mögliche DiGA-Endpunkte entweder basierend auf Ihrer Dokumentation oder basierend auf dem nachträglich mit der DiGA eingeschlagenen Versorgungspfad an und treffen eine sinnvolle Vorabauswahl, damit die Datenauswertung für Ihr Evaluationskonzept zielgerichtet ist und nicht ausufert. Schließlich wollen wir Klarheit und nicht im Trüben fischen.
4. Wie wir Ihnen helfen können
Ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss, klären wir bei medXteam im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung. Das gilt auch für Ihr Evaluationskonzept und Ihre DiGA-Studie!
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung