Die Blog-Reihe zu den Typen klinischer Prüfungen wird im Dezember durch unser „Weihnachtsspezial“ unterbrochen. Hiermit möchten wir Sie umfassend über die wichtigen Änderungen in Bezug auf klinische Prüfungen durch die MDR noch dieses Jahr informieren, damit Sie für 2021 gewappnet sind.
Das Besondere an unserer Aktion ist dabei, dass der Beitrag bis Weihnachten wächst. Jede Woche kommen neue Abschnitte dazu. Im Januar wird es dann mit dem Thema DiGA-Studien weitergehen.
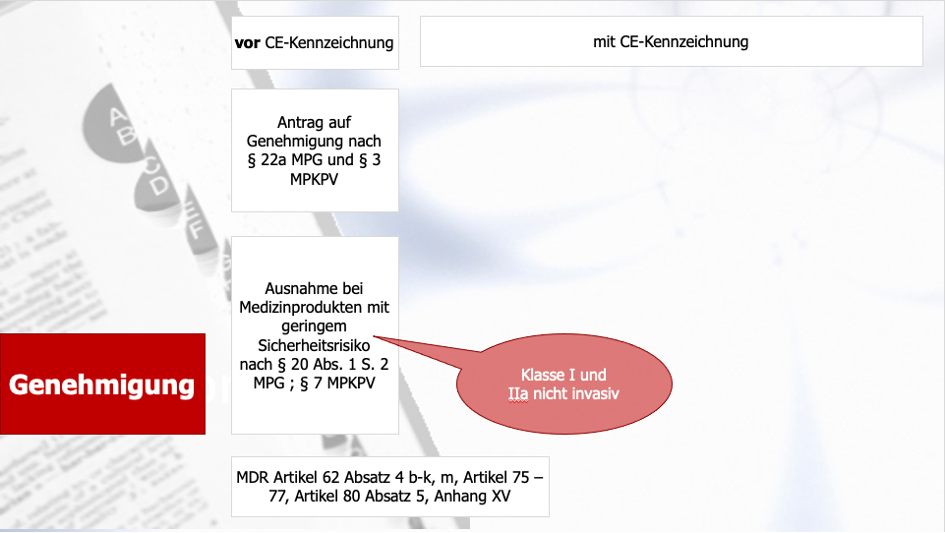
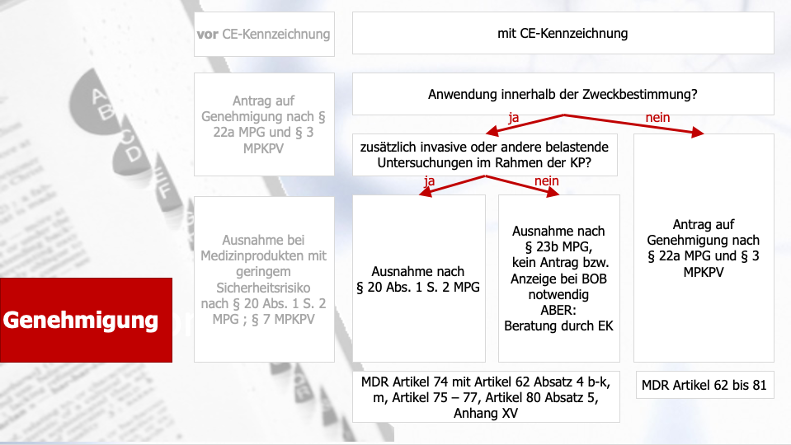
Der erste Teil unseres Dezemberspecials gab Ihnen einen Leitfaden für das Antragsverfahren für klinische Prüfungen im Rahmen des Konformitätsbewertungsverfahrens bei der Bundesoberbehörde und der Ethikkommissionen. Der zweite Teil beschäftigte sich mit dem Antragsverfahren für klinische Prüfungen mit CE-gekennzeichneten Produkten. Der dritte Teil drehte sich um das Antragsverfahren bei sonstigen klinischen Prüfungen. Abschließen werden wir das Weihnachtsspezial heute nun mit dem Thema Safety Reporting bei klinischen Prüfungen, das explizit auch im MDCG-Dokument 2020-10/1 reguliert wird.
Abkürzungen.
BOB (Bundesoberbehörde)
EK (Ethikkommission)
KP (klinische Prüfung)
MDR (medical device regulation; Verordnung 2017/745)
MPEUAnpG (das Medizinprodukte-EU-Anpassungsgesetz wurde am 25.05.2020 vom Bundestag als Gesetze verabschiedet. Dieses MPAnpG-EU beschreibt im Artikel 1 das Medizinprodukte-Durchführungsgesetz (MPDG))
MPDG (das MPDG wird das Medizinproduktegesetz (MPG) ab 26. Mai 2021 schrittweise ablösen und für alle Hersteller und Betreiber von Medizinprodukten in Deutschland rechtsverbindlich sein).
Teil 4: Safety Reporting bei klinischen Prüfungen - Artikel 82 MDR
1. Definitionen
Bei klinischen Prüfungen können jederzeit unerwünschte oder gar schwerwiegende unerwünschte Ereignisse auftreten, von welchen letztere den Behörden zu melden sind. Die MDR definiert ein unerwünschtes Ereignis als
... ein nachteiliges medizinisches Ereignis, eine nicht vorgesehene Erkrankung oder Verletzung oder nachteilige klinische Symptome, einschließlich anormaler Laborbefunde, bei Prüfungsteilnehmern, Anwendern oder anderen Personen im Rahmen einer klinischen Prüfung, auch wenn diese nicht mit dem Prüfprodukt zusammenhängen.
Ein schwerwiegendes unerwünschtes Ereignis wird folgendermaßen definiert:
„schwerwiegendes unerwünschtes Ereignis“ bezeichnet ein unerwünschtes Ereignis, das eine der nachstehenden Folgen hatte:
a) Tod,
b) schwerwiegende Verschlechterung des Gesundheitszustands des Prüfungsteilnehmers, die ihrerseits eine der nachstehenden Folgen hatte:
- lebensbedrohliche Erkrankung oder Verletzung,
- bleibender Körperschaden oder dauerhafte Beeinträchtigung einer Körperfunktion,
- stationäre Behandlung oder Verlängerung der stationären Behandlung des Patienten,
- medizinische oder chirurgische Intervention zur Verhinderung einer lebensbedrohlichen Erkrankung oder Verletzung oder eines bleibenden Körperschadens oder einer dauerhaften Beeinträchtigung einer Körperfunktion,
- chronische Erkrankung,
c) Fötale Gefährdung, Tod des Fötus oder kongenitale körperliche oder geistige Beeinträchtigungen oder Geburtsfehler.
In klinischen Prüfungen definiert man nun noch weitere "Ereignisse":
„Produktmangel“ bezeichnet eine Unzulänglichkeit bezüglich Identifizierung, Qualität, Haltbarkeit, Zuverlässigkeit, Sicherheit oder Leistung eines Prüfprodukts, einschließlich Fehlfunktionen, Anwendungsfehlern oder Unzulänglichkeit der vom Hersteller bereitgestellten Information.
Auch das MDCG-Dokument definiert diese drei Ereignisse in Kapitel 3.
Unerwünschte Ereignisse werden mit "UE" abgekürzt. Im Englischen sind das "Averse events", die Abkürzung dazu laut "AE".
Schwerwiegende unerwünschte Ereignisse werden mit "SUE" abgekürzt. Im Englischen spricht man von "serious Averse events", die mit "SAE" abgekürzt werden.
2. Welche Ereignisse müssen gemeldet werden?
2.1 Klinische Prüfungen nach Artikel 62 der MDR
Zunächst sind in klinischen Prüfungen alle Ereignisse zu dokumentieren. Dazu zählen:
- alle unerwünschten Ereignisse
- alle schwerwiegenden unerwünschten Ereignisse
- alle Produktmängel, die ggf. zu schwerwiegenden unerwünschten Ereignissen hätte führen können
- sowie alle neuen Erkenntnisse zum Produkt in Bezug auf das aufgetretene Ereignis
Welche Ereignisse nun gemeldet werden müssen, geht aus Artikel 80 der MDR und dem MDCG-Dokument hervor:
Der Sponsor meldet unverzüglich über das in Artikel 73 genannte elektronische System allen Mitgliedstaaten, in denen die klinische Prüfung durchgeführt wird,
a) jedes schwerwiegende unerwünschte Ereignis, das einen Kausalzusammenhang mit dem Prüfprodukt, dem Komparator oder dem Prüfverfahren aufweist oder bei dem ein Kausalzusammenhang durchaus möglich erscheint,
b) jeden Produktmangel, der bei Ausbleiben angemessener Maßnahmen oder eines Eingriffs oder unter weniger günstigen Umständen zu schwerwiegenden unerwünschten Ereignissen hätte führen können,
c) alle neuen Erkenntnisse in Bezug auf ein Ereignis gemäß den Buchstaben a und b.
Meldepflichtige Ereignisse müssen vom Sponsor der klinischen Prüfung, der der Hersteller, der gesetzliche Vertreter oder eine andere Person6 oder Einrichtung sein kann, gemeldet werden.
Meldepflichtige Ereignisse müssen zur gleichen Zeit an alle Behörden gemeldet werden, bei denen die klinische Prüfung begonnen wurde. Dazu ist eine Auflistung in der im MDCG-Dokument vorgegebenen Tabelle vorzunehmen.
Die Fristen zur Meldung werden insbesondere in MDCG-2020/1 in Kapitel 8 definiert. Der Sponsor meldet jedes meldepflichtige Ereignis an die Behörden, in deren Zuständigkeitsbereich die klinische Prüfung durchgeführt wird (auch in anderen EU-Ländern und in Drittländern),
- jedes meldepflichtige Ereignis das auf eine unmittelbare Todesgefahr, eine schwere Verletzung oder einer schwere Erkrankung hinweist und das sofortige Abhilfemassnahmen für andere Patienten/Probanden, Anwender oder andere Personen erfordert oder neue Erkenntnis hierzu: unverzüglich, jedoch nicht später als als 2 Kalendertage, nachdem der Sponsor Kenntnis von einem neuen meldepflichtigen Ereignis oder von neuen Informationen im Zusammenhang mit einem bereits gemeldeten Ereignis erlangt hat. Dies schließt signifikante und unerwartete Ereignisse ein, die eine potenzielle Gefahr für die öffentliche Gesundheit darstellen können. Eingeschlossen ist auch die Möglichkeit des Auftretens mehrerer Todesfälle in kurzen Abständen.
- Alle anderen meldepflichtigen Ereignisse oder eine neue Erkenntnis/Aktualisierung dazu: unverzüglich, jedoch nicht später als 7 Kalendertage nach dem Datum, an dem der Sponsor von dem neuen meldepflichtigen Ereignis oder von neuen Informationen im Zusammenhang mit einem bereits gemeldeten Ereignis Kenntnis erlangt hat.
Damit der Sponsor die Fristen einhalten kann, muss dieser dafür Sorge tragen, dass die Meldung der meldepflichtigen Ereignisse durch den Prüfer an den Sponsor unverzüglich, jedoch nicht später als 3 Kalendertage nach Kenntnisnahme des Ereignisses durch das Prüfpersonal des Prüfzentrums erfolgen kann. Dazu ist ein entsprechendes System einzurichten.
2.2 Klinische Prüfungen nach Artikel 74 der MDR
Bei den klinischen Prüfungen nach dem Inverkehrbringen (PMCF-Studien) gelten gemäß Artikel 80 Abschnitt 5 der MDR die Vigilanz-Bestimmungen der Artikel 87 bis 90 und der nach Artikel 91 erlassenen Rechtsakte.
Hier unterscheidet man zwischen den folgenden „Ereignissen“:
Gemäß MDR bezeichnet ein „Vorkommnis“
… eine Fehlfunktion oder Verschlechterung der Eigenschaften oder Leistung eines bereits auf dem Markt bereitgestellten Produkts, einschließlich Anwendungsfehlern aufgrund ergonomischer Merkmale, sowie eine Unzulänglichkeit der vom Hersteller bereitgestellten Informationen oder eine unerwünschte Nebenwirkung.
Ein
… „schwerwiegendes Vorkommnis“ bezeichnet ein Vorkommnis, das direkt oder indirekt eine der nachstehenden Folgen hatte, hätte haben können oder haben könnte: a) den Tod eines Patienten, Anwenders oder einer anderen Person, b) die vorübergehende oder dauerhafte schwerwiegende Verschlechterung des Gesundheitszustands eines Patienten, Anwenders oder anderer Personen, c) eine schwerwiegende Gefahr für die öffentliche Gesundheit.
Gemäß Artikel 87 Abschnitt 1 der MDR ist
… jedes schwerwiegende Vorkommnis im Zusammenhang mit Produkten, die auf dem Unionsmarkt bereitgestellt werden, außer erwarteter Nebenwirkungen, die in den Produktinformationen eindeutig dokumentiert, in der technischen Dokumentation quantifiziert und Gegenstand der Meldung von Trends gemäß Artikel 88 der MDR sind,
zu melden.
Für die schwerwiegenden unerwünschten Ereignisse, bei denen ein Kausalzusammenhang zwischen dem schwerwiegenden unerwünschten Ereignis und dem vorangegangenen Prüfverfahren hergestellt wurde, gelten jedoch die Meldeverfahren für klinische Prüfungen gemäß Artikel 80 der MDR.
Das MDCG-Dokument definiert meldepflichtige Ereignisse bei klinischen Prüfungen im Rahmen des PMCF somit als diejenigen schwerwiegenden unerwünschten Ereignisse, bei denen ein kausaler Zusammenhang zwischen dem schwerwiegenden unerwünschten Ereignis und dem vorausgegangenen Prüfverfahren hergestellt wurde.
Gemäß Artikel 87 der MDR
… hängt die Frist, innerhalb deren die Meldung gemäß Absatz 1 des Artikels 87 zu erfolgen hat, von der Schwere des schwerwiegenden Vorkommnisses ab.
In Abschnitt 3 des Artikels 87 heißt es:
Die Hersteller melden jedes schwerwiegende Vorkommnis im Sinne des Absatzes 1 Buchstabe a unverzüglich, nachdem sie einen Kausalzusammenhang oder einen durchaus möglichen Kausalzusammenhang zwischen dem Vorkommnis und ihrem Produkt festgestellt haben, spätestens jedoch 15 Tage, nachdem sie Kenntnis von dem Vorkommnis erhalten haben.
Abschnitt 4:
Ungeachtet des Absatzes 3 erfolgt im Falle einer schwerwiegenden Gefahr für die öffentliche Gesundheit die Meldung gemäß Absatz 1 unverzüglich, spätestens jedoch zwei Tage, nachdem der Hersteller Kenntnis von dieser Gefahr erhalten hat.
Abschnitt 5:
Ungeachtet des Absatzes 3 erfolgt im Falle des Todes oder einer unvorhergesehenen schwerwiegenden Verschlechterung des Gesundheitszustands einer Person die Meldung unverzüglich, nachdem der Hersteller einen Kausalzusammenhang zwischen dem Produkt und dem schwerwiegenden Vorkommnis festgestellt hat oder sobald er einen solchen Zusammenhang vermutet, spätestens jedoch zehn Tage, nachdem er Kenntnis von dem schwerwiegenden Vorkommnis erhalten hat.
3. Kausalität
Der Zusammenhang zwischen der Anwendung des Medizinprodukts (einschließlich des medizinisch-chirurgischen Verfahrens) und dem Auftreten der einzelnen unerwünschten Ereignisse muss bewertet und kategorisiert werden.
Die Bewertung der Kausalität erfolgt im Rahmen des klinischen Urteilsvermögens des Prüfers. Dabei sind die relevanten Dokumente, wie z. B. das Handbuch des klinischen Prüfers (Investigator’s Brochure, IB), der klinische Prüfplan oder die Risikoanalyse und der Risikomanagementbericht zu Rate zu ziehen. Dort sind nämlich alle vorhersehbaren schwerwiegenden unerwünschten Ereignisse und die potenziellen Risiken aufgelistet und wurden entsprechend bewertet. Das Vorhandensein von Störfaktoren, wie z. B. Begleitmedikation/-behandlung, der natürliche Verlauf der zugrundeliegenden Erkrankung, andere gleichzeitige Erkrankungen oder Risikofaktoren sind ebenfalls zu berücksichtigen.
Die obigen Überlegungen gelten auch für die schwerwiegenden unerwünschten Ereignisse, die in der Kontrollgruppe auftreten.
Jedes schwerwiegende unerwünschte Ereignis wird nach vier verschiedenen Stufen der Kausalität klassifiziert:
1. Kein Zusammenhang
2. Möglicher Zusammenhang
3. Wahrscheinlicher Zusammenhang
4. Kausaler Zusammenhang
Folgende Definitionen finden für die Beurteilung des Zusammenhangs des schwerwiegenden unerwünschten Ereignisses mit dem Prüfprodukt, dem Kontrollprodukt oder dem Prüfverfahren Anwendung:
a. Kein Zusammenhang:
Ein Zusammenhang mit dem Produkt, dem Kontrollprodukt oder dem Prüfverfahren kann ausgeschlossen werden, wenn:
- das Ereignis in keinem zeitlichen Zusammenhang mit der Anwendung des Prüfprodukts oder den mit der Anwendung des Prüfprodukts verbundenen Verfahren steht Prüfproduktes steht,
- das schwerwiegende unerwünschte Ereignis folgt keinem bekannten Reaktionsmuster auf das Medizinprodukt (wenn das Reaktionsmuster zuvor bekannt war) und biologisch unplausibel ist,
- die Beendigung der Anwendung des Medizinprodukts oder die Verringerung des Aktivierungs-/Expositionsgrades - wenn klinisch durchführbar -
und die Wiedereinführung der Anwendung (oder die Erhöhung des Aktivierungs-/Expositionsniveaus) haben keinen Einfluss auf das schwerwiegende unerwünschte Ereignis, - das Ereignis sich auf eine Körperstelle oder ein Organ bezieht, die/das nicht durch das Produkt oder Verfahren beeinflusst werden kann,
- das schwerwiegende unerwünschte Ereignis auf eine andere Ursache zurückgeführt werden kann (z. B. eine zugrundeliegende oder gleichzeitige Krankheit/ein klinischer Zustand, eine Wirkung eines anderen Produkts, Arzneimittels, einer Behandlung oder anderer Risikofaktoren),
- das Ereignis nicht - falls zutreffend - von einem falschen Ergebnis des zur Diagnose verwendeten Prüfprodukts abhängt.
Um den Nicht-Zusammenhang festzustellen, müssen abhängig von der Art des Produkts/Verfahren und des schwerwiegenden unerwünschten Ereignisses möglicherweise nicht alle oben aufgeführten Kriterien gleichzeitig erfüllt werden.
b. Möglicher Zusammenhang:
Der Zusammenhang mit der Anwendung des Prüfprodukts oder des Kontrollprodukts oder der Zusammenhang mit den Verfahren ist schwach, kann aber
kann aber nicht vollständig ausgeschlossen werden. Alternative Ursachen sind ebenfalls möglich (z. B. eine zugrundeliegende oder gleichzeitige Erkrankung/klinischer Zustand oder/und eine Wirkung eines anderen Produkts, Medikaments oder einer Behandlung). Fälle, in denen der Zusammenhang nicht beurteilt werden kann oder keine Informationen vorliegen sollten ebenfalls als möglich eingestuft werden.
c. Wahrscheinlicher Zusammenhang:
Der Zusammenhang mit der Anwendung des Prüfprodukts oder des Kontrollprodukts oder der Zusammenhang mit Verfahren scheint relevant und/oder das Ereignis kann nicht durch eine andere Ursache vernünftig erklärt werden.
d. Kausaler Zusammenhang:
Das schwerwiegende unerwünschte Ereignis steht zweifelsfrei mit dem Prüfprodukt, dem Kontrollprodukt oder mit den Verfahren in Verbindung, wenn:
- das Ereignis eine bekannte Nebenwirkung der Produktkategorie des Prüfprodukts oder ähnlicher Produkte und Verfahren ist,
- das Ereignis in einem zeitlichen Zusammenhang mit der Verwendung/Anwendung des Prüfprodukts oder den Verfahren steht,
- das Ereignis eine Körperstelle oder ein Organ betrifft,
o an der das Prüfprodukt oder die Verfahren angewendet werden
o auf die das Prüfprodukt oder die Verfahren eine Wirkung hat/haben
- das schwerwiegende unerwünschte Ereignis einem bekannten Reaktionsmuster auf das Medizinprodukt folgt (wenn das Reaktionsmuster bereits bekannt ist),
- die Unterbrechung der Anwendung des Medizinprodukts (oder die Verringerung des Aktivierungs-/Expositionsgrades) und die Wiedereinführung seiner Anwendung
(oder Erhöhung des Aktivierungs-/Expositionsniveaus), Auswirkungen auf das schwerwiegende unerwünschte Ereignis (wenn klinisch möglich) hat, - andere mögliche Ursachen (z. B. eine zugrundeliegende oder gleichzeitige Erkrankung/klinischer Zustand oder/und eine Wirkung eines anderen Produkts, Arzneimittels oder einer Behandlung) in angemessener Weise ausgeschlossen werden konnten,
- der Schaden für den Studienteilnehmer auf einen Anwendungsfehler zurückzuführen ist,
- das Ereignis von einem falschen Ergebnis des zur Diagnose verwendeten Prüfprodukts abhängt.
Um den Zusammenhang festzustellen, müssen abhängig von der Art des Produkts/Verfahren und des schwerwiegenden unerwünschten Ereignisses möglicherweise nicht alle oben aufgeführten Kriterien gleichzeitig erfüllt werden.
4. Ausblick
Das war nun Teil 4 unseres „Weihnachtsspezials“ und und gleichzeitig der Abschluss des aufgeteilten Blog-Beitrags zu Änderungen durch die MDR. Auch damit werden wir Sie wieder umfassend über die wichtigen Änderungen in Bezug auf klinische Prüfungen durch die MDR noch dieses Jahr informieren, damit Sie für 2021 gewappnet sind.
Wenn Sie dazu Fragen haben, melden Sie sich gerne. Es steht wie immer unsere kostenlose Erstberatung zur Verfügung. Jetzt verabschiedet sich medXteam aber zunächst in die Weihnachtspause und wünscht allen Lesern ruhige, besinnliche Weihnachtstage. Kommen Sie gesund ins neue Jahr!
Im Januar wird es dann mit dem Thema DiGA-Studien weitergehen.
5. Wie wir Ihnen helfen können
Ob überhaupt und wenn ja welche klinische Prüfung unter welchen Voraussetzungen und gemäß welchen Anforderungen durchgeführt werden muss, klären wir bei medXteam im Rahmen der Pre-Study Phase: In 3 Schritten ermitteln wir die richtige und kosteneffiziente Strategie in Bezug auf die in Ihrem Fall erforderliche klinische Datenerhebung.
Haben Sie jetzt schon erste Fragen?
Eine kostenfreie Erstberatung erhalten Sie hier: kostenlose Erstberatung