The medXteam GmbH blog series continues in the new year and takes up the topic of DiGA studies with the first article in 2021.
Underlying regulations
Digital Healthcare Act (DVG)
Digital Health Applications Ordinance (DiGAV)
DiGA Guide
1. What is a DiGA?
In Chapter 2.1, the guide provides a definition of “digital helpers in the hands of patients”. Accordingly, digital health applications (DiGAs) are “ medical devices of risk class I or IIa (according to the MDR or, as part of the transitional regulations or until the MDR comes into force on May 26, 2021, according to the MDD). This is based
- the main function of DiGA on digital technologies.
- DiGA is not a digital application that is only used to read or control a device; the medical purpose must be achieved essentially through the main digital function.
- The DiGA supports the detection, monitoring, treatment or alleviation of diseases or the detection, treatment, alleviation or compensation of injuries or disabilities.
- The DiGA is not used for primary prevention (see also chapter 2.1.4 DiGA in prevention).
- The DiGA is used jointly by the patient or by the service provider and the patient, i.e. applications that are only used by the doctor to treat the patient (“practice equipment”) are not DiGA.”
DiGA are therefore approved medical products that carry a CE mark and have therefore met the basic safety and performance requirements in accordance with Appendix I of the MDR. However, only Class I and Class IIa medical devices. Also those that are upgraded from class I to class IIa by the MDR. However, all medical devices in classes IIb and III and those that fall under class IIa under Directive 93/42/EEC (MDD) and are classified in class IIb and higher with the MDR do not belong to the group of DiGAs. These cannot be included in the directory.
2 How does DiGA get into the reimbursement register?
The DiGA process is generally only possible with a CE-marked product. The manufacturer can now decide whether he would like to be included in the directory directly and permanently or whether this should initially be done provisionally.
The procedure is designed as a so-called “fast-track procedure”.
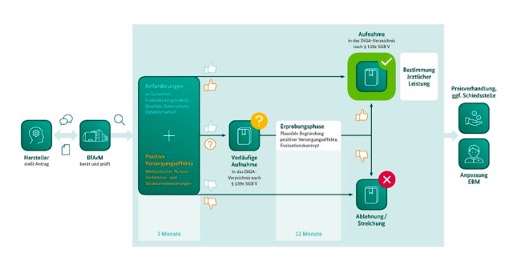
Image1-DiGA: Process of the fast track procedure. Source: DiGA guidelines from BfArM
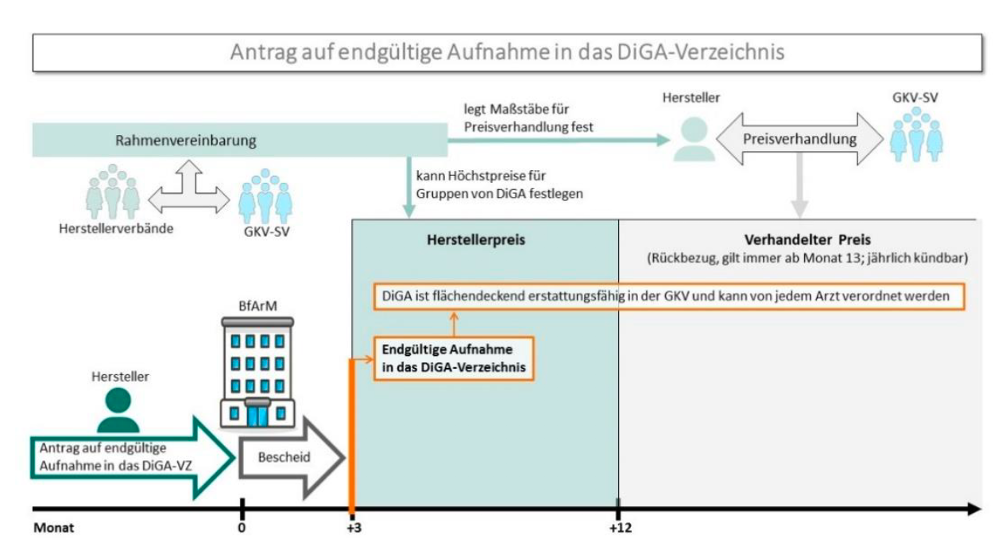
Image2-DiGA: Application for final inclusion in the DiGA directory. Source: DiGA guidelines from BfArM
In order to be included as a DiGA in the reimbursement directory (DiGA directory), various requirements must be met and the review process at the BfArM must be successfully completed. This includes, among other things, an evaluation concept and a clinical study based on it. What does this mean for the medical devices in question? How can the requirements be met and what is the best way to handle the process?
2.1 What is a DiGA study?
In addition to the general requirements
- Safety and functionality
- data protection
- Information security
- Interoperability
and other quality requirements such as:
- robustness
- Consumer protection
- Ease of use
- Support for service providers
- Quality of medical content
- Patient safety
The manufacturer of a DiGA must prove which positive supply effect is achieved. The DiGA guidelines define the positive supply effect as follows:
“As already laid out in the definition of the DiGA in accordance with Section 33a SGB V, the particular focus is on patient-centeredness of the effects to be proven. Both medical benefits and patient-relevant structure and process improvements relate directly to patients and must be proven using appropriate endpoints.”
A medical benefit (mN) is therefore:
- an improvement in the state of health (e.g. reduction of pain, improvement of symptoms, ...),
- a shortening of the duration of the illness (e.g. shortened duration of sick leave, shortened duration of therapy, ...),
- an extension of survival or
- an improvement in the quality of life.
Patient-relevant structure and process improvements (pSVV) are:
- coordination of treatment processes,
- Alignment of treatment with guidelines and recognized standards,
- adherence,
- Facilitating access to care,
- patient safety,
- health literacy,
- patient sovereignty,
- Coping with illness-related difficulties in everyday life
or
- Reduction of therapy-related expenses and burdens on patients and their relatives.
2.2 Requirements for a DiGA study
The legislature places special and clearly defined requirements on a DiGA study. These are described in the DiGA guide :
- In principle, a clinical study must be carried out; publications alone are not enough.
- In this study, the manufacturer must demonstrate at least one positive care effect, which comes either from the area of medical benefit or from the area of patient-relevant structure and process improvements.
- First, the patient group and thus the indications for DiGA for which inclusion in the DiGA directory is requested must be determined. Reimbursement will only be made for these indications. According to the guidelines, the definition and limitation of this patient group must be “based on one or more indications according to ICD-10, whereby only three- and four-digit entries are permitted.”
- The study must be a superiority study because it must show that using DiGA is better than not using it. This is why it is a controlled clinical study: the selection of the comparison or control group must be based on the reality of care. When comparing with treatment without the use of a DiGA, e.g. B. a comparison with standard treatment (the standard of care) is also possible. Or the comparison versus non-treatment is useful if, for example, a DiGA offers care for patients who would otherwise mostly remain untreated and perhaps wait for a place to be treated.
- The study must be a quantitative comparative study and the chosen methodology must be adequate to the chosen subject of investigation. The following designs are possible:
- observational/analytical study: e.g. B. Case/control studies, cohort studies
- experimental intervention study: e.g. B. nonrandomized/randomized controlled trials
- Meta-analyses in the evaluation of our own primary data
- The DiGA study can have a prospective or retrospective approach. The latter, for example, if the medical device has been on the market for a long time and the appropriate data in the required form (comparative) has already been collected with the DiGA and documented accordingly).
- The DiGA study must be carried out in Germany: either as a PMCF study if the medical device is already approved (Article 74 of the MDR or until May 2021: § 23b MPG) or as an approval study to prove the conformity of the medical device with the basic performance - and safety requirements (Article 62 of the MDR or until May 2021: §§ 20 – 23a MPG).
- The DiGA study must still be entered into a study register and the results must be published in full
- The following regulations for clinical trials with medical devices must be applied to the DiGA study:
- DIN EN ISO 14155 “Clinical testing of medical devices on humans – Good Clinical Practice” and the FDA guideline “Design Considerations for Pivotal Clinical Investigations for Medical Devices”
- When medical involvement occurs, the ethical principles of the Declaration of Helsinki apply.
- At least one professional legal consultation must be carried out with an ethics committee (see PMCF study - § 23b MPG!) or under the MDR at least an opinion from the ethics committee must be obtained (Article 74 of the MDR).
This shows the interface to the medical device regulations and the possible use of the clinical data collected in this way for the PMCF (or for the approval of the medical device. It is therefore urgently recommended to comply with ISO 14155 and the MPG/MDR requirements.
3. What we can do for you
A DiGA study is a national specialty, simply because it can only be carried out in Germany. It is also a study requirement for medical devices for which, as part of meeting the basic safety and performance requirements for medical devices, clinical data can normally be waived when demonstrated in the clinical evaluation. Instead, performance data is used.
Basically, we meet DiGA manufacturers where they are and we try to combine regulatory medical device and DiGA requirements with regard to clinical studies as far as possible, since such an effort can certainly be used for both areas. This means you can kill two birds (MDR and DVG) with one stone. That starts e.g. B. when formulating the correct intended purpose of the medical device in order to be able to score points later in negotiations with the health insurance company. It continues with the assessment of the right timing of the DiGA study, with the evaluation concept and study planning and ends with proof of the positive care effect.
That's why we're first working with the DiGA manufacturers to develop a strategy on how they can optimally demonstrate the positive supply effect on their supply path. Depending on your initial situation and your goals.
4. Outlook
In the next blog post we will look in detail at an essential part of the planning phase of a clinical trial, statistical sample size planning.
5. How we can help you
At medXteam we clarify whether and if so which clinical trial needs to be carried out under what conditions and according to what requirements during the pre-study phase: In 3 steps we determine the correct and cost-effective strategy in relation to the clinical trial required in your case Data collection.
Do you already have some initial questions?
You can get a free initial consultation here: free initial consultation