As described in the outlook of the first article in October, the medXteam GmbH blog series now addresses the first type of clinical trials with medical devices: basic research - other clinical trials (MDR Article 82).
Other clinical investigations with medical devices
1 Introduction
Basic research has not only existed since EU Regulation 2017/745 (Medical Device Regulation, MDR), on the contrary. In general, this is about examining feasibility and clarifying fundamental questions. The aim of basic research is therefore:
- research into technical fundamentals
- research into medical principles
- integration into the development of new products
Such activities usually take place in the laboratory:

Figure 1: Feasibility research project
But there are always questions that can only be clarified through tests/studies on humans.
This case was not previously regulated in the MPG. There are only clinical tests with medical devices in accordance with Sections 20 ff.
The MDR now regulates this in Article 82 with the so-called “ other clinical investigations ” , whose implementation at the national level in Germany via the MPEUAnpG (Medical Devices EU Adaptation Act) in Chapter 4, Subsection 2, § 47 to § 61 is detailed.
2. Clarification of terms “medical product” and “prototype”
A medical device is defined in Article 2 of the MDR as follows:
„“Medical device” means an instrument, apparatus, device, software, implant, reagent, material or other item which, according to the manufacturer, is intended for human use and which, alone or in combination, fulfills one or more of the following specific medical purposes should:
-
Diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of diseases,
-
Diagnosing, monitoring, treating, alleviating or compensating for injuries or disabilities,
-
Examination, replacement or modification of anatomy or a physiological or pathological process or condition,
-
Obtaining information through in vitro examination of the human body
-
also from organ, blood and tissue donations
-
from samples and whose intended main effect in or on the human body is not achieved by pharmacological or immunological means or metabolically, but whose mode of action can be supported by such means.“
Fundamental to the definition of a product as a medical device is its intended purpose. According to Article 2 Sentence 12 of the MDR, this is “ … the use for which a product is intended in accordance with the information provided by the manufacturer on the labeling, in the instructions for use or in the advertising or sales material or the advertising or sales information and its information in the clinical evaluation is determined…”
The intended purpose must be formulated as part of the technical documentation by the manufacturer and is decisive for whether a product is simply a product or a medical device. In order for a medical device to be declared as such, it must be able to fulfill its intended purpose under normal conditions of use and correspond to the definition mentioned above.
If the intended purpose of a product does not correspond to the above definition and does not relate to one of the points mentioned above, it is not a medical product. In this case, the MDR requirements for development and initial use do not apply.
What is a medical device and what is not?

Table 1: Distinction between medical device and product
A product referred to as a “prototype” or “demonstrator” is a medical technology product in a very early phase of development. This means that it is a first test version of a new device, procedure or concept that has never been clinically tested. Accordingly, there is no experience with regard to a medical benefit, usability in everyday clinical practice, or the application/use protocol and possible risks associated with the application ".
An initial study with such a prototype/demonstrator is intended to collect initial and basic experience. Furthermore, the feasibility of the new concept/process is to be demonstrated and options for possible development as a medical product are to be made possible. The knowledge and experience gained form the basis on which further development of the prototype/demonstrator into a medical product can take place. This development process according to MDR can then be characterized by numerous changes to the design, equipment, technical functions and configuration in order to achieve sufficient usability and clinical/technical performance. Only a product that meets the demands placed on it in terms of safety and clinical performance can ultimately be placed on the market.
3. Clinical testing on humans – determining the appropriate study type
3.1 Clinical trial to demonstrate the conformity of products (Article 62 MDR) or other clinical trial (Article 82 MDR)
In order to differentiate a clinical trial ( approval study ) according to Article 62 of the MDR from a feasibility study ( other clinical trial ), the objectives of the regulatory requirements within the framework of the MDR must first be presented:
„Based on a high level of health protection for patients and users, this Regulation aims to ensure a smoothly functioning internal market for medical devices, taking into account small and medium-sized enterprises operating in this sector. This regulation also sets high standards for the quality and safety of medical devices, which are intended to address general safety concerns regarding these devices .“
In this context, determining the safety and performance of a medical product plays a fundamental role as a prerequisite for placing it on the market. Placing on the market “ means the initial provision of a product, with the exception of investigational products, on the Union market …” (MDR Article 2, sentence 28). The only exception is the supply of the medical device for the purpose of a clinical trial.
In order for a medical device to be placed on the market, it must have a CE marking. The CE mark indicates that the medical device complies with the legal requirements (is compliant) and that safety and performance have been demonstrated in accordance with the intended purpose under everyday clinical conditions during the evaluation process. The proof is provided by the manufacturer himself and is referred to as the conformity assessment procedure. This involves a clinical assessment of clinical data, the latter being the safety and performance data of the product. In contrast to pharmaceutical law, this data can also be obtained from the literature. The prerequisite is that the relevant data for similar products is available and similarity with your own medical device can be proven. This approach is also known as the literary route.
If this is not possible, performance and safety data must be generated by the manufacturer itself as part of a clinical test - currently the MPG test (§ 20 MPG) or the clinical test to demonstrate the conformity of products (Article 62 MDR). An MPG study is carried out in accordance with the “Regulation for the conduct of clinical trials with medical devices” (MPKPV), which, according to Section 1 (1) of the regulation, is always to be applied when conducting clinical trials in accordance with the MPG, the results of which are used to carry out a conformity assessment procedure in accordance with Medical Devices Regulation (MPV) should be used. After the MDR comes into force, this regulation will be replaced by Regulation (EU) 2017/745 of the European Parliament and of the Council (MDR) and the MPG by the MPEUAnpG (Medical Devices EU Adaptation Act). Clinical trials (Article 62 MDR) and other clinical trials (Article 82 MDR) are then planned and carried out in accordance with the requirements of the MDR and the supplementary national provisions of the MPEUAnpG. In the MDR, too, the focus is on the performance and safety of the medical device during clinical testing in accordance with Article 62:
“Clinical trial” means a systematic investigation involving one or more human subjects and conducted to evaluate the safety or performance of a device.”
The aim of a research project and an associated clinical application should not be to assess the performance and safety of a product in accordance with the current MPG (in conjunction with MPKPV) and MDR as part of a conformity assessment procedure, but rather to assess the medical/technical basis Research that may later be incorporated into the development of new medical products (testing a prototype) does not constitute a “clinical test” within the meaning of the regulations. Although a precise distinction and demarcation is missing in both the MPG and the MDD, this is provided by the MDR, which has been published since May 2017, in Articles 62 and 82:
Article 62: General requirements for clinical trials carried out to demonstrate the conformity of devices
- to determine and verify that a product is designed, manufactured and packaged in such a way that, under normal conditions of use, it is suitable for one or more of the specific purposes listed in Article 2(1) and provides the intended performance as declared ;
- to determine and verify the clinical benefit of a product as claimed by its manufacturer;
- to establish and verify the clinical safety of the device and to determine any undesirable side effects of the device that may occur under normal conditions of use and to assess whether these represent acceptable risks compared to the benefits provided by the device.

Table 2: Objectives and timing of clinical trials to demonstrate product conformity
Article 82: Requirements for other clinical trials (basic research, in-vivo feasibility studies, ...)
- Clinical trials which are not carried out for any of the purposes referred to in Article 62(1) shall comply with the provisions of Article 62(2) and (3), (4)(b), (c), (d), (f), (h) and (l) and paragraph 6.
- In order to protect the rights, safety, dignity and well-being of subjects and to ensure compliance with scientific and ethical principles in clinical trials not carried out for any of the purposes referred to in Article 62(1), each Member State concerned shall: determine appropriate additional requirements for these tests.
And the MPEUAnpG, which comes into force with the MDR, also provides a definition for other clinical trials under Section 3 Sentence 4 Definitions:
"[...] a clinical trial that
-
not part of a systematic and planned product development process or product observation of a current or future manufacturer,
-
carried out with the aim of proving the conformity of a product with the requirements of Regulation (EU) 2017/745,
-
serves to answer scientific or other questions and
-
takes place outside of a clinical development plan in accordance with Annex XIV Part A Number 1 Letter a of Regulation (EU) 2017/745."

Table 3: Other clinical trials
The appropriate study format therefore clearly results from the objective of the clinical trial and the development status of the product to be used.
Therefore, if safety and performance data are to be collected that are used to assess the conformity of an existing prototype (ie the product must also be in an appropriately mature state), the MPG/MDR legal framework applies and a clinical trial must be carried out in accordance with MPKPV/MDR Article 62 are carried out.
On the other hand, if the product is still in an early research stage as a pre-prototype and first of all requires proof of the functionality and usefulness of the underlying concept, initial evidence of the effectiveness of the new process and the feasibility of a subsequent clinical intervention study should therefore be provided is not an MPG study (MDR Article 62), and clinical application does not take place in accordance with the MPKPV. The relevant requirements are described in the following chapter “Conducting a feasibility study”.
Other clinical trials are regulated in Article 82 of the MDR and in Chapter 4, Subsection 2, § 47 to § 61 of the MPEUAnpG (Medical Devices EU Adaptation Act).
3.2 Timing of another clinical trial
Another clinical test is always carried out when it is not intended to evaluate the performance and safety of the medical device. Rather, the focus is on answering scientific or other questions and this can be the case at all times in the “product life cycle”, even before the actual product (i.e. prototype, demonstrator). It may therefore also be the case that “ the other clinical trial is carried out within the scope of the intended purpose covered by the CE marking ” and therefore, it would be about performance/safety and/or benefit in relation to a PMCF study or clinical trial on products that bear the CE marking would be in accordance with Article 74 of the MDR.
3.3 Conducting a feasibility study
The other clinical examination is part of the collection of experience through various procedures for the additional acquisition of knowledge and improvement of specific methods. At the very beginning of such developments, for example, there is an idea that must first be tested for its feasibility, which is usually done within animal models (in vivo studies). In one of these testing phases, initial improvements and adjustments to the original idea take place, which are then tested on animal models. The implementation phase of the idea can include several cycles. The transition to a feasibility study occurs when there is evidence that the idea contributes to improving medical care (Kohnen, 2011).
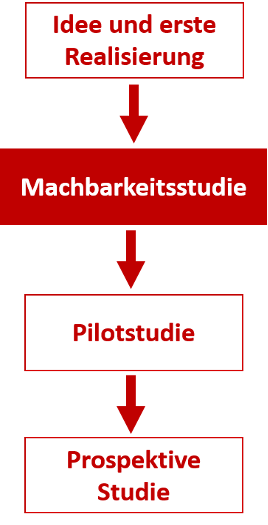
Figure 2: Gaining knowledge in the empirical science of medicine. (Kohnen, 2011)
The aim of such other clinical trials on humans is to provide initial evidence on the effectiveness of a procedure and to examine the feasibility of a possible subsequent clinical intervention study.
If such other clinical trials were not taken into account in the MPG, the MDR now provides a clear definition in Article 82 (see above) and gives clear requirements for this form of clinical trial:
The commissioning of a product is not a legal vacuum, but is subject to extensive safety tests (technical safety, electrical safety, biological safety and standard compliance tests), the so-called “basic requirements” (Appendix I MDD) or “basic safety and performance requirements” (Appendix I MDR) , which must be verified by independent third parties. In addition, a comprehensive risk analysis must be presented and an ethically justifiable benefit-risk balance must be present.
This also follows from Article 82 MDR with reference to Article 62 paragraph 4 letter l:
„(4) A clinical trial in accordance with paragraph 1 can only be carried out if all of the following conditions are met: […]
(l) the investigational device or devices concerned comply with the essential safety and performance requirements set out in Annex I, with the exception of the points covered by the clinical trial; With regard to these points, all precautionary measures have been taken to protect the health and safety of the test participants. This includes, where appropriate, technical and biological safety tests and a pre-clinical assessment as well as provisions in the area of workplace safety and accident prevention, taking into account the latest knowledge .“
Since the other clinical trials are research projects that are part of basic medical research on humans, the general legal regulations and guidelines within medical research also apply. This applies to all other clinical trials, regardless of when they are carried out. To protect patients, users and third parties, a wide variety of requirements must be taken into account. This includes the
- Declaration of Helsinki in its current version.
This means that the ethical standards of medical research on humans are taken into account. Particular attention should be paid to the following points within the project:
- Obtaining informed consent from patients
- Protection of patients unable to consent
- The well-being of the “test subject” is put before the interests of science
- Obtaining a positive vote from the responsible ethics committee This was included in the MDR in Article 82 as follows:
„Clinical trials which are not carried out for one of the purposes referred to in Article 62(1) shall comply with the provisions of Article 62(2) and (3), (4)(b), (c), (d), (f), (h) and (l) and paragraph 6."
This means in particular:
Article 62 paragraph 3
„Clinical trials are designed and conducted in such a way that the protection of the rights, safety, dignity and well-being of the subjects taking part in the trial is guaranteed and takes precedence over all other interests and that the clinical data obtained are scientifically sound, reliable and sound.
Clinical trials are subject to scientific and ethical review. The ethical review is carried out by an ethics committee in accordance with national law. Member States shall ensure that the procedures for review by the ethics committees are compatible with the procedures laid down in this Regulation for the assessment of the application for authorization of a clinical trial. At least one layperson participates in the ethical review .“
Article 62 paragraph 4
„A clinical trial in accordance with paragraph 1 can only be carried out if all of the following conditions are met: […]
(b) an ethics committee established under national law has not issued a negative opinion with regard to the clinical trial, which is valid for the entire territory of the Member State concerned under the national law of the Member State concerned;
(c) the sponsor or its legal representative or a contact person referred to in paragraph 2 is established in the Union;
(d) vulnerable populations and subjects shall be adequately protected in accordance with Articles 64 to 68;
(f) the subject or, if the subject is unable to give informed consent, his or her legal representative has given informed consent in accordance with Article 63;
h) the examinee's right to physical and mental integrity, privacy and protection of his or her personal data in accordance with Directive 95/46/EC is safeguarded.“
In addition, in accordance with Section 48 of the MPEUAnpG, the documents in accordance with Annex XV Chapter II of the MDR must be prepared and submitted to the Ethics Committee.
Subsection 2 with §§ 47 ff in the MPEUAnpG is dedicated to the topic of other clinical trials (requirements, procedures at the ethics committee, etc.) and provides clear national regulations on this.
It is also advisable to consider the application of ISO 14155 for the uniform implementation of a study with medical devices. Although the standard is aimed at clinical testing of medical devices and therefore does not correspond to the intention of a feasibility study, many of the contents can still be applied due to their usefulness. Within the standard, ethical principles have a primary position and Chapter 4 is dedicated to “Ethical Considerations”. Since it is important to protect the safety and well-being of the study participant in accordance with the Declaration of Helsinki (subject insurance, supplementary health care), the standard provides good further assistance in this regard.
The benefit-risk assessment and the justification of a “clinical trial” also play an important role. According to MDR and MPEUAnpG, ethical and safety-related requirements are to be viewed as binding.
For this purpose, ISO 14155 also equates the terms “clinical trial” and “clinical study” with “clinical trial”. To justify a clinical MPG trial, there must be an objective presentation of published and unpublished data as well as a detailed risk analysis and benefit-risk assessment. The justification for a clinical trial is reflected in the trial plan and serves as an evaluation criterion for the ethics committee in its review for a positive assessment. This applies equally to other clinical trials, although due to the early phase and the lack of clinical experience, one can only speak of a benefit-risk assessment with regard to the research project.
4. Outlook
The blog series on clinical trial types will be interrupted in December by our “ Christmas Special .” We would like to inform you comprehensively about the important changes to clinical trials due to the MDR this year so that you are prepared for 2021.
The special thing about our campaign is that the contribution grows until Christmas. New sections with further changes are added every week.
DiGA studies will continue in January.
5. How we can help you
At medXteam we clarify whether and if so which clinical trial needs to be carried out under what conditions and according to what requirements during the pre-study phase: In 3 steps we determine the correct and cost-effective strategy in relation to the clinical trial required in your case Data collection.
Do you already have some initial questions?
You can get a free initial consultation here: free initial consultation