La série de blogs se poursuit cette année pendant l'Avent avec notre « Spécial Avent » en trois parties. Nous souhaitons vous fournir des informations complètes sur les interfaces entre les différents types d’essais cliniques et la documentation technique et les documents pertinents.
La particularité de notre campagne est que la contribution est répartie sur les trois premières semaines de l'Avent. interfaces respectives examinés en détail. Le thème de la gestion des risques dans les essais cliniques se poursuivra en janvier.
La première partie de notre blog spécial présente désormais les interfaces en « étude d'homologation » (essais cliniques conformément à l'article 62 du MDR). La partie 2 met ensuite en avant les études PMCF (Article 74 du MDR, MPDG, ISO 14155) et la troisième et dernière partie explique les interfaces dans les études DiGA .
Abréviations.
BOB (autorité fédérale supérieure)
EK (Commission d'éthique)
KP (examen clinique)
MDR (règlement sur les dispositifs médicaux ; Règlement 2017/745)
MPEUAnpG (la loi d'adaptation européenne aux dispositifs médicaux a été adoptée comme loi par le Bundestag le 25 mai 2020. Ce MPAnpG-EU décrit la loi de mise en œuvre des dispositifs médicaux (MPDG) à l'article 1)
MPDG (la MPDG remplacera progressivement la loi sur les dispositifs médicaux (MPG) à partir du 26 mai 2021 et sera juridiquement contraignante pour tous les fabricants et exploitants de dispositifs médicaux en Allemagne).
Partie 1 : Interfaces avec la documentation technique lors des essais cliniques dans le cadre de la procédure de conformité (études d'approbation) - Article 62, paragraphe 1, du RDM
1. Introduction
Le terme correct pour désigner un essai clinique impliquant des dispositifs médicaux est « essai clinique ».
Une distinction est faite entre les types d'essais cliniques suivants:
- Recherche fondamentale : autres essais cliniques (article 82 du MDR)
- Étude pilote/étude d'approbation : essais cliniques pour démontrer la conformité des produits (article 62 du RIM)
- Etude PMCF : essais cliniques liés aux produits portant le marquage CE (article 74 du MDR)
À cela s’ajoute désormais l’étude dite DiGA, notamment en Allemagne :
- Étudier avec une application numérique de santé (DiGA) pour démontrer les effets positifs des soins afin d'obtenir un statut de remboursement.
- d. R. avec dispositif médical marqué CE : étude PMCF
- Si cela est prévu dans le processus d'approbation, une étude d'approbation est également possible
(Sources : DiGAV, DVG, directives DiGA)
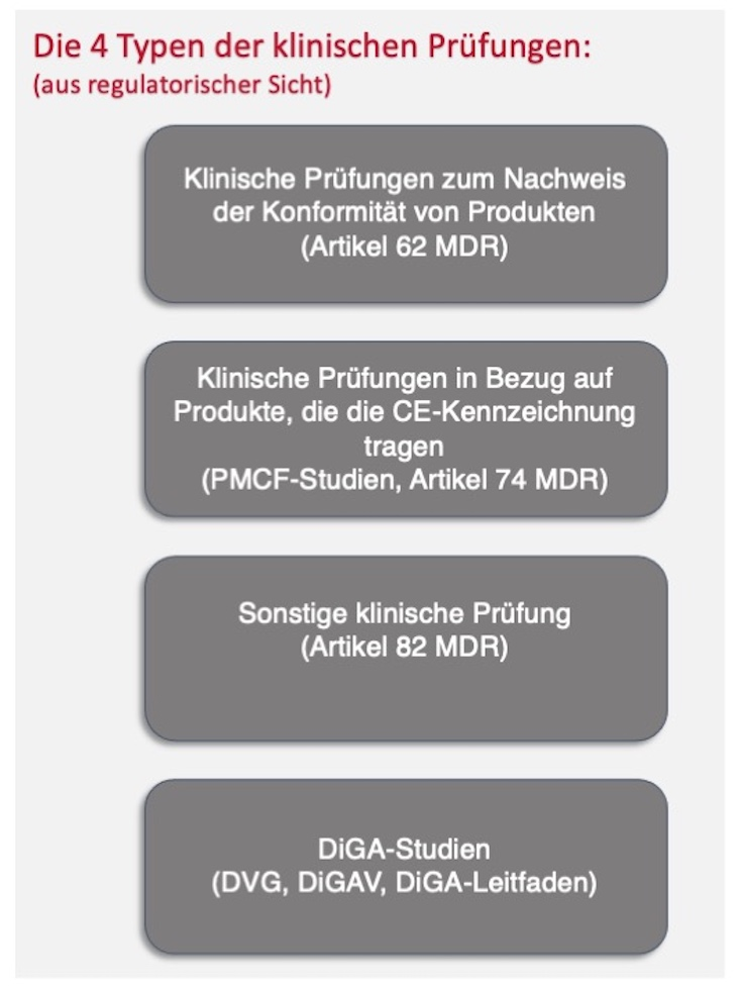
Fig. 1 : Types d'essais cliniques
Ces différents types diffèrent en termes d'exigences réglementaires respectives et donc en termes de différentes interfaces avec la documentation technique du dispositif médical à examiner.
2. Documents à soumettre pour les études d'approbation
Les documents suivants doivent être soumis pour les essais cliniques conformément à l'article 62 du MDR et sont liés à la documentation technique :
- Plan d'essai - pièces jointes selon chapitre annexe XV. II 3 MDR.
- Manuel de l'investigateur clinique - Annexes selon le chapitre de l'annexe XV. II 2 MDR.
- Évaluation préclinique - systèmes selon le chapitre Annexe XV. II 2.3 RDM.
- Mode d'emploi - Annexes conformément à l'Annexe XV Chapitre. II 2.2 RDM.
- Évaluation des risques - investissements conformément au chapitre Annexe XV. II 2,5 ou 4,6 MDR.
- Exigences de base en matière de sécurité et de performance de l'assurance (Exigences de base de l'assurance) - Annexes conformément à l'annexe XV, chapitre II 4.1 du MDR.
- Résultats des tests de sécurité biologique - installations conformément au chapitre Annexe XV. II 2.3 RDM.
- Preuve d'innocuité en matière de sécurité - systèmes conformément à l'annexe XV, chapitre. II 2,5 ou 4,6 MDR.
- Fonctionnement du MP/informations sur le MP (fonctionnement du MP) - systèmes selon l'annexe XV chap. II2.2. MDR.
- Comment ça marche et plus d'informations sur le dispositif médical.
- Analyse et évaluation des risques, y compris les risques résiduels - investissements conformément au chapitre Annexe XV. II 2,5 ou 4,6 MDR.
- Liste des exigences fondamentales de sécurité et de performance - systèmes conformément au chapitre Annexe XV. II 2.7 MDR.
- Si nécessaire, procédés de traitement ou de stérilisation adaptés
- Preuve du marquage CE (Obligatoire si le produit testé porte un marquage CE.)
- Plan d'évaluation clinique - annexes conformément à l'Annexe XV Chapitre II 1.5 MDR ;
- Documentation technique - systèmes conformes à l'annexe XV chapitre II 3.18 et 4.6 MDR sur demande
Le dernier point (annexes conformes à l'annexe XV chapitre II 3.18 et 4.6 MDR) ne doit être soumis que sur demande et comprend :
- Liste des caractéristiques techniques et fonctionnelles du produit, avec référence particulière à celles auxquelles se rapporte l'essai.
- Des détails complets sur la documentation technique disponible, par exemple des documents détaillés d'analyse/gestion des risques ou des rapports d'essais spécifiques, seront fournis sur demande à l'autorité compétente examinant une demande.
2.1 Interfaces vers la documentation technique
Le tableau suivant répertorie les documents mentionnés ci-dessus à soumettre ainsi que les éléments qui y sont contenus et la correspondance respective dans la documentation technique :
|
document |
Exigence réglementaire |
éléments |
Documentation technique |
|
Plan d’étude clinique |
Annexe XV Ch. II 3 MDR |
Etiquetage et description du produit, incluant la destination, le fabricant, la traçabilité, le groupe cible, les matériaux en contact avec le corps humain, les procédures médico-chirurgicales associées à son utilisation ainsi que la formation et l'expérience requises pour son utilisation, trier la littérature de référence, l'état actuel de l'art en matière de soins cliniques dans le domaine concerné |
Description du produit, destination, spécifications du produit, évaluation préclinique comme étape préliminaire de l'évaluation clinique finale avec chapitre sur l'état de la technique, mode d'emploi avec description de l'application |
|
Plan d’étude clinique |
Annexe XV Ch. II 3 MDR |
Risques et bénéfices cliniques du produit testé |
Analyse des risques, rapport de gestion des risques, évaluation préclinique avec évaluation risques-bénéfices |
|
Plan d’étude clinique |
Annexe XV Ch. II 3 MDR |
Informations sur le produit testé, les éventuels comparateurs et autres produits |
Description du produit, mode d'emploi |
|
Plan d’étude clinique |
Annexe XV Ch. II 3 MDR |
caractéristiques techniques et fonctionnelles du produit |
Description du produit, mode d'emploi, spécifications du produit |
|
Manuel du chercheur clinique |
Annexe XV Ch. II 2 MDR |
Étiquetage et description du dispositif, y compris des informations sur l'usage prévu, la classification des risques et la règle de classification applicable conformément à l'annexe VIII, la conception et la fabrication du dispositif et la référence aux générations précédentes et similaires du dispositif. |
Description du produit, destination, spécifications du produit, évaluation préclinique comme étape préliminaire de l'évaluation clinique finale avec chapitre sur l'état de la technique, mode d'emploi avec description de l'application Classification |
|
Manuel du chercheur clinique |
Annexe XV Ch. II 2 MDR |
Les informations du fabricant sur l'installation, l'entretien, le respect des normes d'hygiène et d'utilisation, y compris les exigences de stockage et de manipulation, et, lorsque ces informations sont disponibles, les informations à inclure sur l'étiquette et les instructions d'utilisation à fournir avec le produit lorsqu'il est mis sur le marché. |
|
|
Manuel de l’investigateur clinique/évaluation préclinique |
Annexe XV Ch. II 2.3 RDM |
Évaluation préclinique basée sur les données d'essais et d'expérimentations précliniques pertinents, notamment de calculs de conception, d'essais in vitro, d'essais ex vivo, d'expérimentations animales, d'essais mécaniques ou électriques, d'essais de fiabilité, de validations de stérilisation, de vérifications et validations de logiciels, d'essais de performances, d'évaluations de biocompatibilité et la biosécurité, le cas échéant. |
L'évaluation préclinique comme étape préliminaire de l'évaluation clinique finale |
|
Manuel de l’investigateur clinique/évaluation préclinique |
Annexe XV Ch. II 2.3 RDM |
Données cliniques existantes, en particulier — provenant de la littérature scientifique disponible pertinente sur la sécurité, les performances, les bénéfices cliniques pour les patients, les caractéristiques de conception et la destination du dispositif et/ou de produits similaires ou similaires, — d'autres données cliniques pertinentes disponibles sur la sécurité, les performances, bénéfice clinique pour les patients, caractéristiques de conception et objectif prévu de dispositifs similaires ou similaires du même fabricant, y compris la durée pendant laquelle le dispositif est sur le marché, ainsi que les données issues d'un examen des aspects de performance et de sécurité et de l'utilité clinique et toutes les mesures correctives prises. |
L'évaluation préclinique comme étape préliminaire de l'évaluation clinique finale |
|
Manuel de l’investigateur clinique/évaluation préclinique |
Annexe XV Ch. II 2.3 RDM |
Résumé de l’analyse bénéfice-risque et de la gestion des risques, y compris des informations sur les risques connus ou prévisibles, les éventuels effets secondaires indésirables, les contre-indications et les avertissements. |
Mode d'emploi, évaluation préclinique comme étape préliminaire de l'évaluation clinique finale |
|
Manuel du chercheur clinique |
Annexe XV Ch. II 2 MDR |
Pour les produits contenant un médicament, y compris un dérivé du sang ou du plasma humain, ou pour les produits fabriqués à partir de tissus ou de cellules non viables d'origine humaine ou animale ou de leurs dérivés. |
Seulement dans ce cas : Informations sur le médicament ou les tissus, les cellules ou leurs dérivés et le respect des exigences essentielles de sécurité et de performance pertinentes ainsi que la gestion des risques spécifiques liés au médicament ou aux tissus ou cellules ou leurs dérivés et preuve de l'incorporation de ceux-ci. composants le bénéfice clinique et/ou la sécurité du produit |
|
Manuel du chercheur clinique |
Annexe XV Ch. II 2 MDR |
Une liste détaillant le respect des exigences essentielles de sécurité et de performances pertinentes énoncées à l'annexe I, y compris les normes et spécifications appliquées, en tout ou en partie, et une description des solutions choisies pour répondre aux exigences essentielles de sécurité et de performances pertinentes, le cas échéant. ces normes et spécifications ne sont que partiellement ou pas du tout respectées, voire totalement absentes. |
Liste de contrôle des exigences de base en matière de performances et de sécurité Liste des normes |
|
Mode d'emploi |
Annexe XV Ch. II 2.2 RDM |
-- |
Mode d'emploi |
|
Évaluation des risques |
Annexe XV Ch. II 2.5 ou 4.6 MDR |
Résumé de l’analyse bénéfice-risque et de la gestion des risques, y compris des informations sur les risques connus ou prévisibles, les éventuels effets secondaires indésirables, les contre-indications et les avertissements. Des détails complets sur la documentation technique disponible, par exemple des documents détaillés d'analyse/gestion des risques ou des rapports d'essais spécifiques, seront fournis sur demande à l'autorité compétente examinant une demande. |
Documentation de gestion des risques selon la norme ISO 14971 |
|
Exigences de base en matière de sécurité et de performance des assurances |
Annexe XV Chapitre II 4.1 MDR |
Assurance que les exigences de base en matière de sécurité et de performance sont respectées. |
Liste de contrôle des exigences de base en matière de performances et de sécurité |
|
Résultats des tests de sécurité biologique |
Annexe XV Ch. II 2.3 RDM |
Données issues des tests et essais précliniques pertinents, en particulier les évaluations de biocompatibilité et de biosécurité, le cas échéant. |
Rapports d'essais, rapport de biosécurité |
|
Preuve d'innocuité en matière de sécurité |
Annexe XV Ch. II 2.5 ou 4.6 MDR |
Résumé de l’analyse bénéfice-risque et de la gestion des risques, y compris des informations sur les risques connus ou prévisibles, les éventuels effets secondaires indésirables, les contre-indications et les avertissements. Des détails complets sur la documentation technique disponible, par exemple des documents détaillés d'analyse/gestion des risques ou des rapports d'essais spécifiques, seront fournis sur demande à l'autorité compétente examinant une demande. |
Documentation de gestion des risques selon la norme ISO 14971 |
|
Comment fonctionne le député/informations sur le député |
-- |
Comment ça marche et plus d'informations sur le dispositif médical |
Description du produit, mode d'emploi, spécifications du produit |
|
Analyse et évaluation des risques, y compris les risques résiduels |
Annexe XV Ch. II 2.5 ou 4.6 MDR |
Résumé de l’analyse bénéfice-risque et de la gestion des risques, y compris des informations sur les risques connus ou prévisibles, les éventuels effets secondaires indésirables, les contre-indications et les avertissements. Des détails complets sur la documentation technique disponible, par exemple des documents détaillés d'analyse/gestion des risques ou des rapports d'essais spécifiques, seront fournis sur demande à l'autorité compétente examinant une demande. |
Documentation de gestion des risques selon la norme ISO 14971 |
|
Énumérer les exigences de base en matière de sécurité et de performances |
Annexe XV Ch. II 2.7 RDM |
-- |
Liste de contrôle des exigences de base en matière de performances et de sécurité |
|
Si nécessaire, procédés de traitement ou de stérilisation adaptés |
-- |
Processus de stérilisation, validation |
Documentation du processus de stérilisation |
|
Plan d'évaluation clinique |
Annexe XV Chapitre II 1.5 MDR |
-- |
Plan d'évaluation clinique (CEP) |
|
Documentation technique |
Annexe XV Chapitre II 3.18 et 4.6 MDR sur demande |
Liste des caractéristiques techniques et fonctionnelles du produit. Tous les détails de la documentation technique disponible, par exemple des documents détaillés d'analyse/gestion des risques ou des rapports de tests spécifiques, fournis sur demande. |
Description du produit, mode d'emploi, spécifications du produit Documentation de gestion des risques selon la norme ISO 14971 Rapports de tests de vérification |
Tableau 1 : Documents d'essais cliniques et documentation technique
Le résultat de ce tableau est que les documents suivants issus de la documentation technique doivent être créés avec le dispositif médical final avant le dépôt de la demande d'essai clinique. Le dispositif médical final est donc le produit à tester testé dans l'essai clinique pour lequel les données cliniques sont collectées. C'est à dire. les documents et résultats doivent se référer exactement à ce produit et non à un prototype !
Documentation technique:
- Description du produit
- Utilisation prévue
- spécification de produit
- Plan d'évaluation clinique (CEP)
- Évaluation préclinique comme étape préliminaire de l'évaluation clinique finale avec des chapitres et de la littérature de pointe et une évaluation risques-bénéfices
- Documentation de gestion des risques selon ISO 14971 : PHA, analyse des risques, rapport de gestion des risques
- Mode d'emploi
- Classification
- Liste de contrôle des exigences de base en matière de performances et de sécurité
- Liste des normes
- Rapports de tests de vérification
- Rapport de biosécurité (le cas échéant)
- Documentation du processus de stérilisation (le cas échéant)
Ce n'est que dans le cas de dispositifs médicaux contenant des médicaments ou des tissus, des cellules ou des dérivés que des informations détaillées, qui figurent également dans la documentation technique, doivent également être fournies :
Informations sur le médicament ou les tissus, les cellules ou leurs dérivés et le respect des exigences essentielles de sécurité et de performance pertinentes ainsi que la gestion des risques spécifiques liés au médicament ou aux tissus ou cellules ou leurs dérivés et preuve de l'incorporation de ceux-ci. composants le bénéfice clinique et/ou la sécurité du produit.
2.2 Synergies
Si vous regardez la documentation qui doit être créée avant de postuler à un essai clinique conformément à l'article 62 du MDR, vous remarquerez qu'il s'agit de la quasi-totalité de la documentation technique. Et c'est intentionnel, car le test clinique fait déjà partie de la validation du produit final, les exigences doivent donc déjà avoir été vérifiées. De plus, l’innocuité du produit doit être prouvée avant d’être utilisée sur des humains. C'est à dire. Toutes les exigences de base en matière de performances et de sécurité sont respectées, à l'exception de celles examinées lors de l'essai clinique.
Un bon exemple d'utilisation des synergies lors de la création de documents est le plan de test. De nombreuses sections contiennent le même contenu que dans d'autres documents de documentation technique. L'usage prévu, la description du produit, etc. ne sont que quelques exemples. De plus, les données cliniques sur l'état de l'art sont déjà répertoriées dans une évaluation dite préclinique (un document qui contient les chapitres de l'évaluation clinique à l'exception des données cliniques sur le produit et le PMS etc.), qui sont également utilisées pour le plan de test et le manuel de l'examinateur clinique peuvent être utilisés.
Une telle évaluation préclinique peut également faire partie du manuel de l'investigateur clinique, bien qu'il soit conseillé de créer un document séparé (évaluation préclinique), car cela représente déjà la moitié de la bataille jusqu'à l'évaluation clinique initiale finale.
Et ici, la numérisation des essais cliniques joue à nouveau un rôle :
Avec l'intégration étroite de l'essai clinique non seulement avec le processus de recherche documentaire et donc avec l'évaluation clinique comme rapporté dans le dernier article du blog, mais aussi avec la documentation technique, la numérisation des documents essentiels de l'essai clinique tels que : b.
- Plan d'essai clinique (Annexe XV, Chapitre II, Section 3 du RIM)
- Manuel de l'investigateur clinique (Annexe XV, Chapitre II, Section 2 du MDR)
- évaluation préclinique
possible.
Les avantages de la numérisation sont évidents :
- un travail plus efficace
- Utilisation ciblée des capacités
- Élimination des inefficacités dans la création, la maintenance et la modification du contenu de la documentation technique, de l'évaluation clinique et des recherches documentaires
- réduction à long terme des coûts de soins
Grâce à l'application « Polarion », des interfaces telles que l'objectif, la gestion des risques, la convivialité, l'évaluation clinique, l'essai clinique peuvent être attribuées aux projets et réutilisées si nécessaire. La création et la maintenance des documents sont ainsi considérablement simplifiées et accélérées. De plus, les redondances et les incohérences sont évitées.
3. Perspectives
notre « spécial Avent » portera sur les interfaces vers la documentation technique dans les études PMCF.
4. Comment nous pouvons vous aider
Chez medXteam, nous clarifions si et si oui quel essai clinique doit être réalisé dans quelles conditions et selon quelles exigences pendant la phase préalable à l'étude : En 3 étapes, nous déterminons la stratégie correcte et rentable par rapport à l'essai clinique requis. dans votre cas Collecte de données.
Avez-vous déjà quelques premières questions ?
Vous pouvez obtenir une première consultation gratuite ici : première consultation gratuite