Chez medXteam, l’accent est mis sur les données cliniques. Dans ce contexte, en tant que CRO, nous effectuons non seulement des essais cliniques avec des dispositifs médicaux conformément aux normes MDR et ISO 14155, mais proposons également toutes les autres options et formes de collecte de données. Quelle que soit la forme de collecte de données que vous choisissez, il est important d’avoir une planification solide mais aussi de gérer les différentes options et les exigences respectives. Dans les essais cliniques portant sur des dispositifs médicaux, le promoteur joue un rôle particulièrement important : il est responsable de la bonne planification et de la bonne exécution de l'essai clinique.
Abréviations
Règlement sur les dispositifs médicaux MDR ; Règlement UE 2017/745
Loi de mise en œuvre des dispositifs médicaux MPDG
Loi d'adaptation des dispositifs médicaux MPAnpG
Réglementations sous-jacentes
Règlement UE 2017/745 (MDR)
Loi de mise en œuvre des dispositifs médicaux (MPDG)
ISO 14155
1. Introduction
Les essais cliniques constituent un élément essentiel de la législation relative aux dispositifs médicaux pour garantir la sécurité et les performances des dispositifs médicaux. L'ISO 14155 spécifie les exigences relatives à la réalisation de ces essais cliniques. Dans cet article, nous souhaitons d'abord examiner de plus près le rôle de l'auditeur, puis le rôle important du sponsor et identifier leurs tâches et responsabilités respectives.
Cet article met en lumière les effets du rôle responsable du sponsor et ce qui doit être pris en compte ici - idéalement avant de commencer au centre d'examen.
2. Rôle de l'auditeur
2.1 Définition et nomination
Un enquêteur travaille dans un centre de tests dans le cadre de l'essai clinique. Le chercheur principal nomme cette personne, même si la nomination doit être faite en coordination avec le promoteur. Ceci est crucial car le promoteur doit informer le comité d'éthique des examinateurs et de leurs qualifications lorsqu'il postule à l'examen éthique.
2.2 Tâches de l'examinateur
Le rôle de l'investigateur dans les essais cliniques selon la norme ISO 14155 est d'une importance centrale et est clairement défini par la norme et la loi sur les dispositifs médicaux (MPDG). Des exigences directes et indirectes sont imposées à l'investigateur pour garantir la qualité, l'intégrité et la sécurité des essais cliniques. Certaines des tâches et responsabilités spécifiques de l'auditeur sont :
- Effectuer des procédures cliniques liées aux essais et prendre des décisions clés en matière de traitement clinique et médical liées aux essais.
- Veiller à ce que l'essai clinique soit réalisé conformément au plan d'essai (conformément à l'article 62, paragraphe 1, n° 1 du MPDG).
- Si l'examinateur est un médecin ou un dentiste, il doit effectuer l'information et obtenir le consentement du participant au test (conformément à l'article 28, paragraphe 2, MPDG).
- Participer aux réunions d'enquêteurs organisées par le promoteur.
- Assurer l'exactitude, l'attribution, l'exhaustivité, la lisibilité et l'actualité des données sources ainsi que des données soumises au promoteur dans les CRF (Case Report Forms) et tous les rapports requis.
- Pour les sujets qui interrompent leur participation à l'étude, demandez l'autorisation de collecter des données de suivi sur leur état ou leur maladie.
- Évaluation des événements indésirables (EI), notamment en termes de gravité et de relation avec le produit expérimental.
- En cas de circonstances pouvant affecter la sécurité des participants au test, des utilisateurs ou des tiers, toutes les mesures de sécurité nécessaires doivent être prises immédiatement pour éviter un danger direct ou indirect (conformément à l'article 66, paragraphe 1, MPDG).
- S'il n'y a qu'un seul examinateur dans le centre d'examen, il reprend automatiquement les tâches de l'examinateur principal.
Les responsabilités de l'investigateur sont étendues et variées, chaque étape contribuant à assurer la sécurité des patients et l'intégrité de l'essai clinique. Il est donc essentiel que les auditeurs soient parfaitement formés et connaissent et comprennent toutes les réglementations et exigences pertinentes.
2.3 Indépendance de l'auditeur
Il est essentiel que l'auditeur soit indépendant. Il ne doit ni être influencé par le promoteur ni influencer d'autres personnes ou institutions impliquées dans l'examen.
2.4 Communication avec le sponsor
L'investigateur doit recevoir toutes les informations nécessaires du promoteur pour garantir une évaluation et une documentation cohérentes des résultats obtenus au cours de l'essai.
3. Rôle du sponsor
3.1 Définition et responsabilité
Le promoteur est responsable du lancement, de la gestion et du financement de l'essai clinique. Il doit être basé dans l'Union européenne ou désigner un représentant légal basé dans l'UE. Ce représentant assume la responsabilité du respect des obligations du promoteur et est l'interlocuteur des autorités et du comité d'éthique.
3.2 Responsabilités du sponsor
La norme ISO 14155 définit diverses tâches pour le sponsor, notamment :
- Planification et préparation de l'essai clinique : cela comprend, entre autres, la détermination des besoins de l'essai clinique, la gestion des risques, le développement du concept et la sélection du personnel clinique.
- Réalisation de l'essai clinique : cela comprend la garantie du respect du plan d'essai, du suivi, de la qualité des données et de la protection des données personnelles.
- Évaluation de la sécurité : le promoteur doit enregistrer, évaluer et documenter tous les événements indésirables.
- Arrêt de l'essai clinique : cela inclut également la communication avec les autorités et, si nécessaire, l'interruption ou la fin de l'essai.
Le deuxième point, la conduite de l’essai clinique, joue ici un rôle particulièrement crucial. Pour garantir cela, les mesures suivantes doivent être prises en compte :
A l'avance, avant le départ au centre de test :
- Sélection du bon centre d'essai : Le centre d'essai doit disposer des installations et des ressources nécessaires et, idéalement, avoir déjà une expérience en matière d'essais cliniques.
- Formation du centre de test : Le centre doit être informé des exigences et spécifications en vigueur ainsi que des principes juridiques et doit avoir reçu une formation régulière à cet égard.
- Vérification des qualifications : s'assurer que l'investigateur, ou au moins l'investigateur principal d'un centre multi-évaluateurs, possède une formation GCP-MDR à jour.
- Qualifications de l'équipe d'étude : L'équipe réalisant l'étude, en particulier les assistants d'étude (infirmières de l'étude), doivent être correctement qualifiés et formés. Une formation régulière peut aider à maintenir les connaissances à jour.
- Audits préliminaires : des audits indépendants peuvent être menés avant le début de l'étude pour vérifier la conformité aux directives GCP.
- Canaux de communication clairs : des procédures claires de communication et de reporting doivent être établies avant le début de l’étude.
Pendant l'étude :
- Surveillance régulière : pendant la conduite de l'étude, le site d'étude doit être surveillé régulièrement pour garantir que les protocoles de l'étude sont correctement suivis. Cela se fait via une surveillance, également stipulée dans la norme ISO 14155.
- Audits internes : le site d'essai peut effectuer des audits internes pour garantir lui-même la conformité aux politiques et procédures de l'étude. Mais le sponsor peut également réaliser un audit sur place pour s’assurer de la bonne mise en œuvre.
- Exigences en matière de documentation : tous les documents pertinents doivent être enregistrés et archivés correctement et rapidement.
- Formation continue : en cas de modification de la réglementation ou du protocole d'étude, l'ensemble de l'équipe d'étude doit être recyclé.
Selon l'étude :
- Visite de clôture : Une fois l'étude terminée, le rendez-vous de contrôle final est effectué conformément à la norme ISO 14155 pour vérifier le respect de toutes les exigences et la bonne mise en œuvre sur site.
- Boucle de rétroaction : les erreurs ou les problèmes survenus au cours de l'étude doivent être analysés et intégrés dans la formation et les processus futurs.
Le respect de ces étapes peut garantir le bon déroulement des essais cliniques sur un site d’essai.
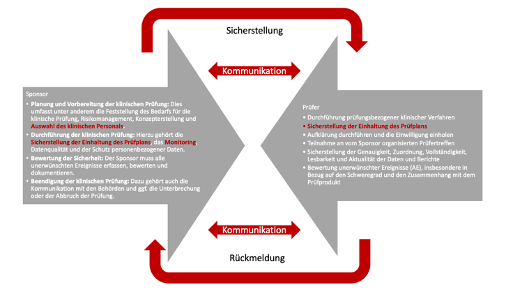
Figure : Sponsor de l'interaction - examinateur
4. Conclusion
En résumé, l'investigateur et le promoteur jouent un rôle central dans les essais cliniques ISO 14155. Leurs rôles et responsabilités respectifs sont clairement définis pour garantir l’intégrité et la qualité des essais cliniques. Il est extrêmement important que les deux parties remplissent leur rôle correctement et avec diligence pour garantir la sécurité et l'efficacité des dispositifs médicaux pour les patients. En particulier avant une étude, une préparation et une planification ciblées peuvent grandement contribuer à sa mise en œuvre réussie. Cela comprend, entre autres, une formation complète du centre d'essai pour garantir que non seulement l'investigateur mais l'ensemble de l'équipe d'étude sont formés conformément aux BPC-MDR. L'identification et la qualification précoces des infirmières de l'étude et des autres membres du personnel clé du site d'étude peuvent également contribuer de manière décisive à minimiser les éventuels obstacles ou retards au cours de l'étude. De plus, une stratégie de communication claire entre le sponsor et le centre de test doit être établie au préalable afin d'éviter dès le départ les malentendus et les sources potentielles d'erreurs. Compte tenu des exigences élevées et de l’énorme responsabilité qu’impliquent les essais cliniques, une préparation proactive et bien pensée est essentielle au succès. Il est de la responsabilité conjointe du sponsor et de l'auditeur de garantir que toutes les exigences et normes sont non seulement respectées, mais également mises en œuvre de manière cohérente.
5. Comment nous pouvons vous aider
Chez medXteam, nous clarifions si et si oui quel essai clinique doit être réalisé dans quelles conditions et selon quelles exigences pendant la phase préalable à l'étude : En 3 étapes, nous déterminons la stratégie correcte et rentable par rapport à l'essai clinique requis. dans votre cas Collecte de données.
Si un essai clinique doit être réalisé, les exigences fondamentales de sécurité et de performance doivent d’abord être respectées. Les données de l’essai clinique alimentent ensuite l’évaluation clinique, qui à son tour constitue la base des activités de suivi clinique post-commercialisation (PMCF) (y compris une étude PMCF).
De plus, tous les fabricants de dispositifs médicaux exigent un système de gestion de la qualité (QMS), y compris lors du développement de produits de classe I.
Nous vous accompagnons tout au long de votre projet avec votre dispositif médical, depuis une première consultation gratuite, une aide à l'introduction d'un système QM, la planification et la mise en œuvre de l'étude jusqu'à la documentation technique - toujours avec une référence primaire aux données cliniques du produit : de du début à la fin Fin.
Avez-vous déjà quelques premières questions ?
Vous pouvez obtenir une première consultation gratuite ici : première consultation gratuite